ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ - ą║ą╗ą░čüąĖčćąĮąĖ ąĘąĮą░ą║ąŠą▓ąĖ, ą║ą░ąŠ ąĮą░čüą╗ąĄčÆąĄąĮą░ ąĖ ą┤ąĖčśąĄč鹥čéčüą║ą░ č鹥čĆą░ą┐ąĖčśą░
ąĪą░ą┤čƹȹ░čś
- 1ąÜą░ą║ąŠ čüąĄ ą╝ą░ąĮąĖč乥čüčéčāčśąĄ ą▒ąŠą╗ąĄčüčé č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░
- 2ą£ąĄčģą░ąĮąĖąĘą╝ čĆą░ąĘą▓ąĖčéąĖą░ ąŠą▒čüąĄčüčéą▓ą░
- 3ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖą░ ą┐čĆąĖ ą┤ąĄčåą░
- 4ąĪąĖą╝ą┐č鹊ą╝ąĖ ąŠą▒ąŠą╗ąĄčÜą░
- 5ąÆąĘčĆąŠą║ąĖ ąĖ ą┐čĆąŠą▓ąŠą║ą░čéąĖą▓ąĮąĖąĄ čäą░ą║č鹊čĆąĖ
- 6ąöąĖą░ą│ąĮąŠčüčéąĖčåčü
- 7ąøąĄč湥čÜąĄ ą║ą╗ą░čüąĖčćąĮąĄ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖąĄ
- 8ąśčüčģčĆą░ąĮą░ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ ąĖ ą┤ąĖčśąĄčéą░ą╗ąĮą░ č鹥čĆą░ą┐ąĖčśą░
- 9ą¤čĆąĄčģčĆą░ąĮą░ ąĘą░ ą┐čĆąĄą┤čłą║ąŠą╗čüą║čā ąĖ čłą║ąŠą╗čüą║čā ą┤ąĄčåčā
- 10ą¤čĆąŠąĖąĘą▓ąŠą┤čüčéą▓ąŠ ąĮą░ ą¤ąÜąŻ
- 11ąÜą░ą║ąŠ ą║ąŠąĮčéčĆąŠą╗ąĖčüą░čéąĖ ąĮąĖą▓ąŠ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā ą║čĆą▓ąĖ
- 12ąÆąĖą┤ąĄąŠ
ąæąŠą╗ąĄčüčé, čćąĖčśą░ čśąĄ ą┐ąŠčśą░ą▓ą░ ą┐ąŠą▓ąĄąĘą░ąĮą░ čüą░ ą┤ąĄč乥ą║čéąĖą╝ą░ ą│ąĄąĮąĄčéčüą║ąŠą│ čøąĄą╗ąĖčśčüą║ąŠą│ ą░ą┐ą░čĆą░čéą░ - č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ - čāą║čÖčāč湥ąĮą░ čśąĄ čā ą╝ą░ą╗čā ą╗ąĖčüčéčā ąĮą░čüčÖąĄą┤ąĮąĖčģ ą▒ąŠą╗ąĄčüčéąĖ ą║ąŠčśąĄ čüąĄ ą╗ąĖčśąĄč湥. ą×čéą║čĆąĖą▓ą░čć ąŠą▓ąĄ ą▒ąŠą╗ąĄčüčéąĖ ą▒ąĖąŠ čśąĄ ąĮąŠčĆą▓ąĄčłą║ąĖ ą╗ąĄą║ą░čĆ ąśąÉ ążąĄą╗ą╗ąĖąĮą│, ą║ą░čüąĮąĖčśąĄ čśąĄ ąŠčéą║čĆąĖą▓ąĄąĮąŠ ą┤ą░ čĆą░ąĘą▓ąŠčś ąĖ č鹊ą║ ą▒ąŠą╗ąĄčüčéąĖ ąŠą┤ą│ąŠą▓ą░čĆą░ čśąĄą┤ąĮąŠą╝ ą│ąĄąĮčā ą║ąŠčśąĖ čüąĄ ąĘąŠą▓ąĄ ą│ąĄąĮ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ čģąĖą┤čĆąŠą║čüąĖą╗ą░ąĘąĄ (ą┤čāą│ąĖ čĆą░ą╝ 12-č鹊ą│ čģčĆąŠą╝ąŠąĘąŠą╝ą░ ą║ąŠčśąĖ čüą░ą┤čƹȹĖ ą┤ąŠ 4,5% čāą║čāą┐ąĮąŠą│ ąöąØąÜ čøąĄą╗ąĖčśčüą║ąŠą│ ą╝ą░č鹥čĆąĖčśą░ą╗ą░). ąØą░čüą╗ąĄčÆąĄąĮąĖ ą┤ąĄč乥ą║čé ą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ ą┤ąĄą╗ąĖą╝ąĖčćąĮąĄ ąĖą╗ąĖ ą┐ąŠčéą┐čāąĮąĄ ą┤ąĄąĘą░ą║čéąĖą▓ą░čåąĖčśąĄ ąĄąĮąĘąĖą╝ą░ čśąĄčéčĆąĄ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ-4-čģąĖą┤čĆąŠą║čüąĖą╗ą░ąĘąĄ.
ąÜą░ą║ąŠ čüąĄ ą╝ą░ąĮąĖč乥čüčéčāčśąĄ ą▒ąŠą╗ąĄčüčé č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░

ą£ąĄčģą░ąĮąĖąĘą╝ čĆą░ąĘą▓ąĖčéą║ą░ ąŠą▒čüąĄčüčéą▓ą░
ąśąĮčģąĖą▒ąĖčåąĖčśą░ ą╝ąĄčéą░ą▒ąŠą╗ąĖčéą░ čāąĘčĆąŠą║ąŠą▓ą░ąĮą░ ą╝čāčéą░čåąĖčśčüą║ąŠą╝ ąĖąĮą░ą║čéąĖą▓ą░čåąĖčśąŠą╝ ąĄąĮąĘąĖą╝ą░ čśąĄ ą░ą║čéąĖą▓ą░čåąĖčśą░ ą┐ąŠą╝ąŠčøąĮąĖčģ ą┐čāč鹥ą▓ą░ ąĘą░ čĆą░ąĘą╝ąĄąĮčā č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░. ąÉčĆąŠą╝ą░čéčüą║ą░ ą░ą╗čäą░-ą░ą╝ąĖąĮąŠ ą║ąĖčüąĄą╗ąĖąĮą░, ą║ą░ąŠ čĆąĄąĘčāą╗čéą░čé ąĮąĄąĖčüą┐čĆą░ą▓ąĮąĖčģ ą┐čĆąŠčåąĄčüą░ čĆą░ąĘą╝ąĄąĮąĄ, čĆą░ąĘą╗ą░ąČąĄ čüąĄ ąĮą░ č鹊ą║čüąĖčćąĮąĄ ą┤ąĄčĆąĖą▓ą░č鹥 ą║ąŠčśąĖ čüąĄ ąĮąĄ č乊čĆą╝ąĖčĆą░čśčā ą┐ąŠą┤ ąĮąŠčĆą╝ą░ą╗ąĮąĖą╝ čāčüą╗ąŠą▓ąĖą╝ą░:
- č乥ąĮąĖą╗ą┐ąĖčĆčāą▓ąĖčćąĮą░ ą║ąĖčüąĄą╗ąĖąĮą░ (č乥ąĮąĖą╗ą┐ąĖčĆčāą▓ą░čé) - ą╝ą░čüąĮąĖ ą░čĆąŠą╝ą░čéčüą║ąĖ ą░ą╗čäą░-ą║ąĄč鹊ą░čåąĖą┤, čÜąĄąĮąŠ ąŠą▒čĆą░ąĘąŠą▓ą░čÜąĄą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ ą╝ąĖčśąĄą╗ąĖąĮąĖąĘą░čåąĖčśąĄ ą┐čĆąŠčåąĄčüą░ ąĮąĄčāčĆąŠąĮą░ ąĖ ą┤ąĄą╝ąĄąĮčåąĖčśąĄ;
- č乥ąĮąĖą╗ą╝ą░ą║čéąĖčćąĮą░ ą║ąĖčüąĄą╗ąĖąĮą░ - ą┐čĆąŠąĖąĘą▓ąŠą┤ ą║ąŠčśąĖ ąĮą░čüčéą░čśąĄ č鹊ą║ąŠą╝ čĆąĄą│ąĄąĮąĄčĆą░čåąĖčśąĄ č乥ąĮąĖą╗ ą▓ąĖčĆąĖąŠąĮčüą║ąĄ ą║ąĖčüąĄą╗ąĖąĮąĄ;
- ążąĄąĮąĖą╗ąĄčéąĖą╗ą░ą╝ąĖąĮ - ą┐ąŠč湥čéąĮąŠ čśąĄą┤ąĖčÜąĄčÜąĄ ąĘą░ ą▒ąĖąŠą╗ąŠčłą║ąĖ ą░ą║čéąĖą▓ąĮąĄ ą┐čĆąĄąĮąŠčüąĮąĖą║ąĄ ąĄą╗ąĄą║čéčĆąŠą║ąĄą╝ąĖčśčüą║ąĖčģ ąĖą╝ą┐čāą╗čüą░, ą┐ąŠą▓ąĄčøą░ą▓ą░ ą║ąŠąĮčåąĄąĮčéčĆą░čåąĖčśčā ą┤ąŠą┐ą░ą╝ąĖąĮą░, ą░ą┤čĆąĄąĮą░ą╗ąĖąĮą░ ąĖ ąĮąŠčĆąĄą┐ąĖąĮąĄčäčĆąĖąĮą░;
- ą×čĆčéčģąŠčäčģąĄąĮąĖą╗ą░čåąĄčéą░č鹥 - č鹊ą║čüąĖčćąĮą░ čüčāą┐čüčéą░ąĮčåą░ ą║ąŠčśą░ čāąĘčĆąŠą║čāčśąĄ ą║čĆčłąĄčÜąĄ ą╝ąĄčéą░ą▒ąŠą╗ąĖčćą║ąĖčģ ą┐čĆąŠčåąĄčüą░ č乥ą║ą░ą╗ąĮąĖčģ čüą┐ąŠčśąĄą▓ą░ čā ą╝ąŠąĘą│čā.
ą£ąĄą┤ąĖčåąĖąĮčüą║ą░ čüčéą░čéąĖčüčéąĖą║ą░ ą┐ąŠą║ą░ąĘčāčśąĄ ą┤ą░ čśąĄ ą┐ą░č鹊ą╗ąŠčłą║ąĖ ąĖąĘą╝ąĄčÜąĄąĮąĖ ą│ąĄąĮ ą┐čĆąĖčüčāčéą░ąĮ čā 2% ą┐ąŠą┐čāą╗ą░čåąĖčśąĄ, ą░ą╗ąĖ čüąĄ ąĮąĄ ą╝ą░ąĮąĖč乥čüčéčāčśąĄ. ąōąĄąĮąĄčéčüą║ąĖ ą┤ąĄč乥ą║čé čüąĄ ą┐čĆąĄąĮąŠčüąĖ ąĮą░ ą┤ąĄč鹥 ąŠą┤ čĆąŠą┤ąĖč鹥čÖą░ čüą░ą╝ąŠ čā ą┐čĆąĖčüčāčüčéą▓čā ą▒ąŠą╗ąĄčüčéąĖ ą║ąŠą┤ ąŠą▒ą░ ą┐ą░čĆčéąĮąĄčĆą░, ą░ ą▒ąĄą▒ą░ čā 50% čüą╗čāčćą░čśąĄą▓ą░ ą┐ąŠčüčéą░čśąĄ ąĮąŠčüąĖą╗ą░čå ą╝čāčéąĖčĆą░ąĮąŠą│ ą│ąĄąĮą░, ąŠčüčéą░čśčāčøąĖ ąĘą┤čĆą░ą▓ąĖ. ąÆąĄčĆąŠą▓ą░čéąĮąŠčøą░ ą┤ą░ čøąĄ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ ą║ąŠą┤ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ ą┤ąŠą▓ąĄčüčéąĖ ą┤ąŠ ą▒ąŠą╗ąĄčüčéąĖ čśąĄ 25%.
ąÜąŠčśąĖą╝ čéąĖą┐ąŠą╝ čüąĄ ąĮą░čüą╗ąĄčÆčāčśąĄ
ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čā ą┤ąĄčåąĄ
ąÜą╗ą░čüąĖčćąĮąĖ ąŠą▒ą╗ąĖą║ ą│ąĄąĮąĖčéą░ą╗ąĮąĖčģ ą▒ąŠą╗ąĄčüčéąĖ ą║ąŠą┤ ą┤čśąĄčåąĄ čüąĄ čā ą▓ąĄčøąĖąĮąĖ čüą╗čāčćą░čśąĄą▓ą░ ą╝ą░ąĮąĖč乥čüčéčāčśąĄ čüą┐ąŠčÖą░ čāąŠčćčÖąĖą▓ąĖą╝ ąĘąĮą░ą║ąŠą▓ąĖą╝ą░, ąŠą┤ 3-9 ą╝čśąĄčüąĄčåąĖ ąČąĖą▓ąŠčéą░. ąØąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ čüą░ ą┤ąĄč乥ą║čéąĮąĖą╝ ą│ąĄąĮąŠą╝ ąĖąĘą│ą╗ąĄą┤ą░čśčā ąĘą┤čĆą░ą▓ąŠ, ą░ ą┐ąŠčüąĄą▒ąĮąŠčüčé čśąĄ čüą┐ąĄčåąĖčäąĖčćąĮą░ ąĮą░ą▓ąĖą║ą░ (ąĖąĘą│ą╗ąĄą┤) ą┤ąĄč鹥čéą░. ąĪąĖą╝ą┐č鹊ą╝ čüąĄ čśą░ą▓čÖą░ 6-12 ą╝ąĄčüąĄčåąĖ ąĮą░ą║ąŠąĮ čĆąŠčÆąĄčÜą░.
ą¤ąÜąŻ čéąĖą┐ ąśąś ą║ą░čĆą░ą║č鹥čĆąĖčłąĄ čćąĖčÜąĄąĮąĖčåą░ ą┤ą░ čüąĄ ą┐čĆą▓ąĖ ą║ą╗ąĖąĮąĖčćą║ąĖ čüąĖą╝ą┐č鹊ą╝ąĖ čśą░ą▓čÖą░čśčā ąĮą░ą║ąŠąĮ 1,5 ą│ąŠą┤ąĖąĮąĄ ąŠą┤ čéčĆąĄąĮčāčéą║ą░ ą┐ąŠč湥čéą║ą░ čüą▓ąĄčéą╗ąŠčüčéąĖ. ąŚąĮą░čåąĖ ą▒ąŠą╗ąĄčüčéąĖ ąĮąĄ ąĮąĄčüčéą░čśčā ąĮą░ą║ąŠąĮ ą┤ąĖčśą░ą│ąĮąŠčüčéąĖą║ąŠą▓ą░čÜą░ ą│ąĄąĮąĄčéčüą║ąĖčģ ą░ą▒ąĮąŠčĆą╝ą░ą╗ąĮąŠčüčéąĖ ąĖ ą┐ąŠč湥čéą║ą░ ą┤ąĖčśąĄčéą░ą╗ąĮąĄ č鹥čĆą░ą┐ąĖčśąĄ. ą×ą▓ą░čś čéąĖą┐ čāčĆąŠčÆąĄąĮąĖčģ ą▒ąŠą╗ąĄčüčéąĖ č湥čüč鹊 ą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ čäą░čéą░ą╗ąĮąŠą│ ąĖčüčģąŠą┤ą░ 2-3 ą│ąŠą┤ąĖąĮąĄ ąČąĖą▓ąŠčéą░ ą┤čśąĄč鹥čéą░. ąØą░čśč湥čłčøąĖ čüąĖą╝ą┐č鹊ą╝ąĖ čéąĖą┐ą░ ąśąś ą¤ąÜąŻ čüčā:
- ąĖąĘčĆą░ąČąĄąĮą░ ąŠą┤čüčéčāą┐ą░čÜą░ čā ą╝ąĄąĮčéą░ą╗ąĮąŠą╝ čĆą░ąĘą▓ąŠčśčā;
- čģąĖą┐ąĄčĆčĆąĄčäą╗ąĄą║čüąĖčśą░;
- ą║čĆčłąĄčÜąĄ ą╝ąŠč鹊čĆąĖčćą║ąĖčģ čäčāąĮą║čåąĖčśą░ čüą▓ąĖčģ ąĄą║čüčéčĆąĄą╝ąĖč鹥čéą░;
- čüąĖąĮą┤čĆąŠą╝ ąĮąĄą║ąŠąĮčéčĆąŠą╗ąĖčüą░ąĮąĖčģ ą║ąŠąĮčéčĆą░ą║čåąĖčśą░ ą╝ąĖčłąĖčøą░.
ąÜą╗ąĖąĮąĖčćą║ąĖ ąĘąĮą░čåąĖ ą╝čāčéą░čåąĖčśčüą║ąĖčģ ą┐čĆąŠą╝ąĄąĮą░ čā ą│ąĄąĮąĖą╝ą░ čéąĖą┐ą░ ąśąśąś čüčā čüą╗ąĖčćąĮąĖ ą▒ąŠą╗ąĄčüčéąĖą╝ą░ ą║ąŠčśąĖ čüąĄ čśą░ą▓čÖą░čśčā čā čéąĖą┐čā ąśąś. ąØąĄą┤ąŠčüčéą░čéą░ą║ č鹥čéčĆą░čģąĖą┤čĆąŠą▒ąĖąŠą┐č鹥čĆąĖąĮą░ ą║ą░čĆą░ą║č鹥čĆąĖčłąĄ čéčĆąĖčśą░ą┤ą░ čüą┐ąĄčåąĖčäąĖčćąĮąĖčģ čüąĖą╝ą┐č鹊ą╝ą░:
- ą▓ąĖčüąŠą║ čüč鹥ą┐ąĄąĮ ą╝ąĄąĮčéą░ą╗ąĮąĄ čĆąĄčéą░čĆą┤ą░čåąĖčśąĄ;
- ą┐čĆąĖą▓ąĖą┤ąĮąŠ čüą╝ą░čÜąĄąĮčā ą▓ąĄą╗ąĖčćąĖąĮčā ą╗ąŠą▒ą░čÜąĄ čā ąŠą┤ąĮąŠčüčā ąĮą░ ą┤čĆčāą│ąĄ ą┤ąĄą╗ąŠą▓ąĄ č鹥ą╗ą░;
- čüą┐ą░čüčéąĖčćąĮąŠčüčé ą╝ąĖčłąĖčøą░ (čāąĘ ą╝ąŠą│čāčøąĮąŠčüčé ą┐ąŠčéą┐čāąĮąŠą│ ą│čāą▒ąĖčéą║ą░ ą┐ąŠą║čĆąĄčéą░ ąĄą║čüčéčĆąĄą╝ąĖč鹥čéą░).

ą£ą░ąĮąĖč乥čüčéą░čåąĖčśąĄ ą▒ąŠą╗ąĄčüčéąĖ čüąĄčćąĖą▓ą░
ąóąŠą║ąŠą╝ ą║ą╗ąĖąĮąĖčćą║ąĖčģ čüčéčāą┤ąĖčśą░ ąĖ ąĘą░ą┐ą░ąČą░čÜą░, čüčāą│ąĄčĆąĖčüą░ąĮąŠ čśąĄ ą┤ą░ čśąĄ ąĄč乥ą║ą░čé ą▒ąĖąŠąóąŠą║čüąĖčćąĮąĖ ą┤ąĄčĆąĖą▓ą░čéąĖ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čĆą░ąĘą╝ąĄąĮąĄ čāąĘčĆąŠą║čāčśčā čüą╝ą░čÜąĄčÜąĄ ąĖąĮč鹥ą╗ąĄą║čéčāą░ą╗ąĮąĖčģ čüą┐ąŠčüąŠą▒ąĮąŠčüčéąĖ, čłč鹊 čśąĄ ą┐čĆąŠą│čĆąĄčüąĖą▓ąĮąŠ ąĖ ą╝ąŠąČąĄ ą┤ąŠą▓ąĄčüčéąĖ ą┤ąŠ ą┤ąĄą╝ąĄąĮčåąĖčśąĄ (ąŠą╗ąĖą│ąŠčäčĆąĄąĮąĖčśą░, ąĖą┤ąĖąŠčéąĖ). ą£ąĄčÆčā ą▓čśąĄčĆąŠą▓ą░čéąĮąĖą╝ čāąĘčĆąŠčåąĖą╝ą░ ąĖčĆąĄą▓ąĄčƹʹĖą▒ąĖą╗ąĮąĖčģ ą┐ąŠą▓čĆąĄą┤ą░ ą╝ąŠąČą┤ą░ąĮąĄ ą░ą║čéąĖą▓ąĮąŠčüčéąĖ, čüą╝ą░čéčĆą░ čüąĄ ą┤ą░ čśąĄ ąĮą░čśčĆą░ąĘčāą╝ąĮąĖčśą░ ą┐ąŠčüčÖąĄą┤ąĖčåą░ čüą╝ą░čÜąĄčÜą░ ąĮąĖą▓ąŠą░ čéąĖčĆąŠąĘąĖąĮą░, ąĮąĄą┤ąŠčüčéą░čéą║ą░ ąĮąĄčāčĆąŠčéčĆą░ąĮčüą╝ąĖč鹥čĆą░ ą║ąŠčśąĖ ą┐čĆąĄąĮąŠčüąĄ ąĖą╝ą┐čāą╗čüąĄ ąĖąĘą╝ąĄčÆčā ąĮąĄčāčĆąŠąĮą░.
ąóą░čćąĮą░ čāąĘčĆąŠčćąĮąŠ-ą┐ąŠčüčÖąĄą┤ąĖčćąĮą░ ą▓ąĄąĘą░ ąĖąĘą╝ąĄčÆčā ąĮą░čüčÖąĄą┤ąĮąĖčģ ą▒ąŠą╗ąĄčüčéąĖ ąĖ ą┐ąŠčĆąĄą╝ąĄčøą░čśą░ ą╝ąŠąĘą│ą░ ą┤ąŠ čüą░ą┤ą░ ąĮąĖčśąĄ ąŠčéą║čĆąĖą▓ąĄąĮą░, ą║ą░ąŠ ąĖ ą╝ąĄčģą░ąĮąĖąĘą░ą╝ čĆą░ąĘą▓ąŠčśą░ ąĘą▒ąŠą│ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśąĄ čéą░ą║ą▓ąĖčģ ą╝ąĄąĮčéą░ą╗ąĮąĖčģ čüčéą░čÜą░ ą║ą░ąŠ čłč鹊 čüčā ąĄčģą┐čĆą░ą║čüąĖčśą░, ąĄčģąŠą╗ą░ą╗ąĖčśą░, ą┤ąĖą▓čÖąĖ ąĮą░ą┐ą░ą┤ąĖ ąĖ čĆą░ąĘą┤čĆą░ąČčÖąĖą▓ąŠčüčé. ąĀąĄąĘčāą╗čéą░čéąĖ ą░ąĮą░ą╗ąĖąĘą░ čāą║ą░ąĘčāčśčā ąĮą░ č鹊 ą┤ą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ ąĖą╝ą░ ą┤ąĖčĆąĄą║čéą░ąĮ č鹊ą║čüąĖčćąĮąĖ ąĄč乥ą║ą░čé ąĮą░ ą╝ąŠąĘą░ą║, čłč鹊 čéą░ą║ąŠčÆąĄ ą╝ąŠąČąĄ čāąĘčĆąŠą║ąŠą▓ą░čéąĖ čüą╝ą░čÜąĄčÜąĄ ąĖąĮč鹥ą╗ąĖą│ąĄąĮčåąĖčśąĄ.ąĪą┐ąĄčåąĖčäąĖčćąĮąŠčüčéąĖ čüčéčĆčāą║čéčāčĆąĄ ąĖ č乥ąĮąŠčéąĖą┐ą░
ąĪ ąŠą▒ąĘąĖčĆąŠą╝ ą┤ą░ ąĘą░čüąĖčøąĄąĮąŠčüčé ą┐ąĖą│ą╝ąĄąĮčéą░ ą║ąŠąČąĄ ąĖ ą┤ą╗ą░ą║ąĄ ąĘą░ą▓ąĖčüąĖ ąŠą┤ ąĮąĖą▓ąŠą░ čéąĖčĆąŠąĘąĖąĮą░ čā ą╝ąĖč鹊čģąŠąĮą┤čĆąĖčśą░ą╝ą░ čģąĄą┐ą░č鹊čåąĖčéą░, ą░ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ ą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ ąĘą░čāčüčéą░ą▓čÖą░čÜą░ ą║ąŠąĮą▓ąĄčƹʹĖčśąĄ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░, ą┐ą░čåąĖčśąĄąĮčéąĖ čüą░ ąŠą▓ąŠą╝ ą▒ąŠą╗ąĄčłčøčā ąĖą╝ą░čśčā č乥ąĮąŠčéąĖą┐čüą║ąĄ ą║ą░čĆą░ą║č鹥čĆąĖčüčéąĖą║ąĄ (čĆąĄčåąĄčüąĖą▓ąĮąĖ ąĘąĮą░čåąĖ). ą¤ąŠą▓ąĄčøą░ąĮąĖ č鹊ąĮčāčü ą╝ąĖčłąĖčøą░ čāąĘčĆąŠą║čāčśąĄ ą┐ąŠčśą░ą▓čā ą┤ąĄą▓ąĖčśą░čåąĖčśą░ čā čüčéčĆčāą║čéčāčĆąĖ č鹥ą╗ą░ - ą┐ąŠčüčéą░čśąĄ ą┤ąĖčüą┐ą╗ą░čüčéąĖčćą░ąĮ. ą×ą┤ą╗ąĖčćąĮąĄ čüą┐ąŠčÖąĮąĄ ą║ą░čĆą░ą║č鹥čĆąĖčüčéąĖą║ąĄ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čāą║čÖčāčćčāčśčā:
- čģąĖą┐ąŠą┐ąĖą│ą╝ąĄąĮčéą░čåąĖčśą░ - čüą▓ąĄčéą╗ą░ ą║ąŠąČą░, ą▒ą╗ąĄą┤ąŠ ą┐ą╗ą░ą▓ąĄ ąŠčćąĖ, ą▒ąĄąĘą▒ąŠčśąĮą░ ą║ąŠčüą░;
- čåąĖąĮąĖąĘą░ą╝ čāą┤ąŠą▓ą░;
- čüą╝ą░čÜąĄąĮą░ ą▓ąĄą╗ąĖčćąĖąĮą░ ą│ą╗ą░ą▓ąĄ;
- čüą┐ąĄčåąĖčäąĖčćą░ąĮ ą┐ąŠą╗ąŠąČą░čś č鹥ą╗ą░ - ą║ą░ą┤ą░ ą┐ąŠą║čāčłą░ą▓ą░č鹥 ą┤ą░ čüč鹊蜹Ėč鹥 ąĖą╗ąĖ čüąĄą┤ąĖč鹥 ą┤ąĄč鹥 čāąĘąĖą╝ą░ ą┐ąŠą╗ąŠąČą░čś "ą║čĆąŠčśą░čćą░" (čĆčāą║ąĄ ąĖ ąĮąŠą│ąĄ čüą░ą▓ąĖčśąĄąĮąĄ čā ąĘą│ą╗ąŠą▒ąŠą▓ąĖą╝ą░).
ąĪąĖą╝ą┐č鹊ą╝ąĖą▒ąŠą╗ąĄčüčé
ą¤čĆą░ą▓ąŠą▓čĆąĄą╝ąĄąĮąŠ, ążąĄą╗ą╗ąĖąĮą│ąŠą▓ą░ ą▒ąŠą╗ąĄčüčé čüąĄ čāčüą┐ąĄčłąĮąŠ čéčĆąĄčéąĖčĆą░ ą┐čĆąĖą╗ą░ą│ąŠčÆą░ą▓ą░čÜąĄą╝ ąĖčüčģčĆą░ąĮąĄ ąĖ čĆą░ąĘą▓ąŠčś ą┤ąĄč鹥čéą░ čüąĄ ąŠą┤ą▓ąĖčśą░ čā čüą║ą╗ą░ą┤čā čüą░ čÜąĄą│ąŠą▓ąŠą╝ čüčéą░čĆąŠčüąĮąŠą╝ ą│čĆčāą┐ąŠą╝. ąóąĄčłą║ąŠčøą░ čā ąŠčéą║čĆąĖą▓ą░čÜčā ą╝čāčéą░čåąĖčśąĄ ą│ąĄąĮą░ ą╗ąĄąČąĖ čā čćąĖčÜąĄąĮąĖčåąĖ ą┤ą░ čüąĄ čĆą░ąĮąĖ ąĘąĮą░čåąĖ č鹥賹║ąŠ ąŠčéą║čĆąĖą▓ą░čśčā čćą░ą║ ąĖ ą║ąŠą┤ ąĖčüą║čāčüąĮąŠą│ ą┐ąĄą┤ąĖčśą░čéčĆą░. ą×ąĘą▒ąĖčÖąĮąŠčüčé čüąĖą╝ą┐č鹊ą╝ą░ ą║ąŠąĮą│ąĄąĮąĖčéą░ą╗ąĮąĄ ą▒ąŠą╗ąĄčüčéąĖ čüąĄ ą┐ąŠą▓ąĄčøą░ą▓ą░ ą║ą░ą║ąŠ ą┤ąĄč鹥 čĆą░čüč鹥, čśąĄčĆ čāą┐ąŠčéčĆąĄą▒ą░ ą┐čĆąŠč鹥ąĖąĮčüą║ąĄ čģčĆą░ąĮąĄ ą┤ąŠą┐čĆąĖąĮąŠčüąĖ čĆą░ąĘą▓ąŠčśčā ą”ąØąĪ ą┐ąŠčĆąĄą╝ąĄčøą░čśą░.
ąŚąĮą░ą║ąŠą▓ąĖ ą║ąŠą┤ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ
ąĪą░ ą┐ąŠč湥čéą║ąŠą╝ ą┤ąŠčśąĄčÜą░, ąĮąŠą▓ąĖ ą┐čĆąŠč鹥ąĖąĮąĖ ą┐ąŠčćąĖčÜčā ą┤ą░ čāą╗ą░ąĘąĄ čā č鹥ą╗ąŠ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ čüą░ ą╝ą╗ąĄą║ąŠą╝, čłč鹊 čüą╗čāąČąĖ ą║ą░ąŠ ą║ą░čéą░ą╗ąĖąĘą░č鹊čĆ ąĘą░ ą┐ąŠčśą░ą▓čā ą┐čĆą▓ąĖčģ ąĘąĮą░ą║ąŠą▓ą░, čłč鹊 čśą░čüąĮąŠ čāą║ą░ąĘčāčśąĄ ąĮą░ č鹊 ą┤ą░ čśąĄ ą▒ąŠą╗ąĄčüčé ą┐ąŠč湥ą╗ą░ ą┤ą░ ąĮą░ą┐čĆąĄą┤čāčśąĄ. ąĪą┐ąĄčåąĖčäąĖčćąĮąĄ ą║ą╗ąĖąĮąĖčćą║ąĄ ą╝ą░ąĮąĖč乥čüčéą░čåąĖčśąĄ ą▒ąŠą╗ąĄčüčéąĖ čāą║čÖčāčćčāčśčā:
- čéčĆą░čśąĮąŠ ą┐ąŠą▓čĆą░čøą░čÜąĄ (č湥čüč鹊 čāąĘąĄč鹊 ąĘą░ čāčĆąŠčÆąĄąĮąŠ čüčāąČą░ą▓ą░čÜąĄ ą│ąŠą╗ą╝ą░ąĮą░);
- č湥čüčéą░ ą┤ąĖčüą╗ąŠą║ą░čåąĖčśą░;
- ąĮąĄą╝ą░ čĆąĄą░ą║čåąĖčśąĄ ąĮą░ čüą┐ąŠčÖą░čłčÜąĄ ą┐ąŠą┤čĆą░ąČą░čśąĄ;
- ą╝ąĖčłąĖčøąĮą░ ą┤ąĖčüč鹊ąĮąĖčśą░ (čüą╝ą░čÜąĄąĮą░ ąĮą░ą┐ąĄč鹊čüčé ą╝ąĖčłąĖčøą░);
- ą║ąŠąĮą▓čāą╗ąĘąĖą▓ąĮąĖ čüąĖąĮą┤čĆąŠą╝ (ąĄą┐ąĖą╗ąĄą┐čéąĖčćą║ąĖ ąĖą╗ąĖ ąĄą┐ąĖą╗ąĄą┐čéąĖčćą║ąĖ ąĮą░ą┐ą░ą┤ą░čśąĖ).
ąĪąĖą╝ą┐č鹊ą╝ąĖ ą║ąŠą┤ ą┤ąĄčåąĄ ą┐ąŠčüą╗ąĄ 6 ą╝ąĄčüąĄčåąĖ
ąÉą║ąŠ ą╝ą░ąĮąĖč乥čüčéą░čåąĖčśą░ ą│ąĄąĮąĄčéčüą║ąĄ ą▒ąŠą╗ąĄčüčéąĖ ąĮąĖčśąĄą┤ąŠ {ą╗ąŠ (ąĖą╗ąĖ ąĮąĖčśąĄ ą▓ąĖ | ąĄąĮąŠ) č鹊ą║ąŠą╝ ą┐čĆą▓ąĖčģ 6 ą╝ąĄčüąĄčåąĖ ąŠą┤ čĆąŠ | ąĄčÜą░ ą┤ąĄč鹥čéą░, ą┐ą░ ąĮą░ą║ąŠąĮ ąŠą▓ąŠą│ ą┐ąĄčĆąĖąŠą┤ą░ ą▓ąĄ} ą╝ąŠ╩╗ąĄč鹥 ą┐čĆąĄčåąĖąĘąĮąŠ ąŠą┤čĆąĄą┤ąĖčéąĖ ąĘą░ąŠčüčéą░čéą░ą║ ą┐čüąĖčģąŠą╝ąŠč鹊čĆąĮąŠą│ čĆą░ąĘą▓ąŠčśą░. ąĪąĖą╝ą┐č鹊ą╝ąĖ ą│ąĄąĮąĄčéčüą║ąĖčģ ą┐ąŠčĆąĄą╝ąĄčøą░čśą░ čāąĘčĆąŠą║ąŠą▓ą░ąĮąĖčģ ąĮąĄą┤ąŠčüčéą░čéą║ąŠą╝ ąĄąĮąĘąĖą╝ą░ ą║ąŠą┤ ą┤ąĄčåąĄ čüčéą░čĆąĖčśąĄ ąŠą┤ čłąĄčüčé ą╝ąĄčüąĄčåąĖ čüčā:
- čüą╝ą░čÜąĄčÜąĄ ą░ą║čéąĖą▓ąĮąŠčüčéąĖ (ą┤ąŠ ą┐ąŠčéą┐čāąĮąĄ ąĖąĮą┤ąĖč乥čĆąĄąĮčéąĮąŠčüčéąĖ);
- ąĮąĄą┤ąŠčüčéą░čéą░ą║ ą┐ąŠą║čāčłą░čśą░ ą┤ą░ čāčüčéą░ąĮąĄ, čüąĄą┤ąĄ;
- ą┐ąŠčüąĄą▒ą░ąĮ "ą╝ąĖčłčśąĖ" ą╝ąĖčĆąĖčü ą║ąŠąČąĄ (ą╝ąĖčĆąĖčü ą┐ą╗ąĖčśąĄčüąĮąĖ ąĮą░čüčéą░čśąĄ ąĘą▒ąŠą│ ą┐ąŠą▓ą╗ą░č湥čÜą░ č鹊ą║čüąĖčćąĮąĖčģ ą┤ąĄčĆąĖą▓ą░čéą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ ą║čĆąŠąĘ ąČą╗ąĖčśąĄąĘą┤ąĄ ąĘąĮąŠčśąĮąĖčåąĄ ąĖ čāčĆąĖąĮ);
- ą│čāą▒ąĖčéą░ą║ čüą┐ąŠčüąŠą▒ąĮąŠčüčéąĖ ą▓ąĖąĘčāą░ą╗ąĖąĘą░čåąĖčśąĄ ą╗ąĖčåą░ čĆąŠą┤ąĖč鹥čÖą░;
- čÖčāčłč鹥čÜąĄ ą║ąŠąČąĄ;
- ą┐ąŠčśą░ą▓ą░ ą┤ąĄčĆą╝ą░čéąĖčéąĖčüą░, ąĄą║čåąĄą╝ą░, čüą║ą╗ąĄčĆąŠą┤ąĄčĆą╝ąĄ.
ą¤čĆąŠą│čĆąĄčüąĖčśą░ ą▒ąŠą╗ąĄčüčéąĖ čā ąŠą┤čüčāčüčéą▓čā ą╗ąĄč湥čÜą░ čā ą┤ąĄčéąĖčÜčüčéą▓čā
ąÉą║ąŠ čĆą░ąĘą▓ąŠčśąĮą░ ąŠą┤čüčéčāą┐ą░čÜą░ ąĮąĖčüčā ąŠčéą║čĆąĖą▓ąĄąĮą░ čā ą┤čśąĄčéąĖčÜčüčéą▓čā ąĖ ąĮąĖčśąĄ ą┐čĆąŠą▓ąĄą┤ąĄąĮ ąŠą┤ą│ąŠą▓ą░čĆą░čśčāčøąĖ čéčĆąĄčéą╝ą░ąĮ, ą▒ąŠą╗ąĄčüčé ą┐ąŠčćąĖčÜąĄ ą░ą║čéąĖą▓ąĮąŠ ąĮą░ą┐čĆąĄą┤ąŠą▓ą░čéąĖ ąĖ č湥čüč鹊 ą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ ąĖąĮą▓ą░ą╗ąĖą┤ąĖč鹥čéą░. ąØąĄą┤ąŠčüčéą░čéą░ą║ č鹥čĆą░ą┐ąĖčśąĄ čā čĆą░ąĮąŠčś čäą░ąĘąĖ ą▒ąŠą╗ąĄčüčéąĖ čāąĘčĆąŠą║čāčśąĄ ą┐ąŠčśą░ą▓čā čéą░ą║ą▓ąĖčģ čüąĖą╝ą┐č鹊ą╝ą░ ą▒ąŠą╗ąĄčüčéąĖ čā ą┤ąŠą▒ąĖ ąŠą┤ 1,5 ą│ąŠą┤ąĖąĮą░:
- ą╝ąĖą║čĆąŠčåąĄčäą░ą╗ąĖčśą░ (čüą╝ą░čÜąĄąĮą░ ą▓ąĄą╗ąĖčćąĖąĮą░ ą╝ąŠąĘą│ą░);
- ąŠčéąĖčåą░čÜąĄ (ą┐ąŠą╝ąĄčĆą░čÜąĄ ą│ąŠčĆčÜąĄą│ čĆąĄą┤ą░ ąĘčāą▒ą░ ąĮą░ą┐čĆąĄą┤);
- ą║ą░čüąĮąĖčśąĄ ąĄčĆčāą┐čåąĖčśąĄ ąĘčāą▒ą░;
- čģąĖą┐ąŠą┐ą╗ą░ąĘąĖčśą░ ą│ą╗ąĄčÆąĖ (ą╝čĆčłą░ą▓ąŠčüčé ąĖą╗ąĖ ą┐ąŠčéą┐čāąĮąŠ ąŠą┤čüčāčüčéą▓ąŠ ąĘčāą▒ąĮąĄ čåą░ą║ą╗ąĖąĮąĄ);
- ąŠą┤ą╗ą░ą│ą░čÜąĄ ą╗ąĖąĮą│ą▓ąĖčüčéąĖčćą║ąŠą│ čĆą░ąĘą▓ąŠčśą░ ą┤ąŠ ą┐ąŠčéą┐čāąĮąŠą│ ąĮąĄą┤ąŠčüčéą░čéą║ą░ ą│ąŠą▓ąŠčĆą░;
- 3, 4 čüč鹥ą┐ąĄąĮ ąŠą╗ąĖą│ąŠčäčĆąĄąĮąĖčśąĄ (ą╝ąĄąĮčéą░ą╗ąĮą░ čĆąĄčéą░čĆą┤ą░čåąĖčśą░, ą╝ąĄąĮčéą░ą╗ąĮą░ čĆąĄčéą░čĆą┤ą░čåąĖčśą░);
- čāčĆąŠčÆąĄąĮąĄ čüčĆčćą░ąĮąĄ ą▒ąŠą╗ąĄčüčéąĖ (ą┤ąĄč乥ą║čéąĖ čā čüčéčĆčāą║čéčāčĆąĖ čüčĆčćą░ąĮąŠą│ ą╝ąĖčłąĖčøą░, čüčĆčåąĄ,ą▓ąĄą╗ąĖą║ąĄ ą┐ąŠčüčāą┤ąĄ);
- ą┐ąŠčĆąĄą╝ąĄčøą░čśąĖ ą▓ąĄą│ąĄčéą░čéąĖą▓ąĮąŠą│ čüąĖčüč鹥ą╝ą░ (ą░ą║čĆąŠčåąĖčśą░ąĮąŠąĘą░, ą┐ąŠčśą░čćą░ąĮąŠ ąĘąĮąŠčśąĄčÜąĄ, ą░čĆč鹥čĆąĖčśčüą║ą░ čģąĖą┐ąŠč鹥ąĮąĘąĖčśą░);
- ą║ąŠąĮčüčéąĖą┐ą░čåąĖčśą░.
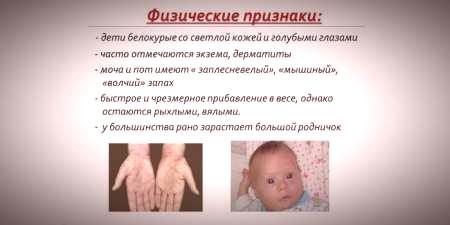
ąÆąĘčĆąŠą║ąĖ ąĖ ą┐čĆąŠą▓ąŠą║ą░čéąĖą▓ąĮąĖąĄ čäą░ą║č鹊čĆąĖ
ąÉčéąĖą┐ąĖčćąĮąĖ ąŠą▒ą╗ąĖčåąĖ ą▒ąŠą╗ąĄčüčéąĖ ą╝ąŠą│čā čüąĄ ąĖąĘą╗ąĄčćąĖčéąĖ ą┐čĆąĖą╗ą░ą│ąŠčÆą░ą▓ą░čÜąĄą╝ ąĖčüčģčĆą░ąĮąĄ. ą×ą▓ą░ ąŠą┤čüčéčāą┐ą░čÜą░ čüčā ą┐ąŠčüą╗ąĄą┤ąĖčåą░ ąĮąĄą┤ąŠčüčéą░čéą║ą░ č鹥čéčĆą░čģąĖą┤čĆąŠą┐č鹥čĆąĖąĮą░, ą┤ąĄčģąĖą┤čĆąŠą┐č鹥čĆąĖąĮ čĆąĄą┤čāą║čéą░ąĘąĄ (čĆąĄčéą║ąŠ - ą┐ąĖčĆą▓ą░č鹥č鹥čéčĆą░čģąĖą┤čĆąŠą┐č鹥čĆąĖąĮčüčéą░ąĮąĘą░, ą│ą▓ą░ąĮąŠąĘąĖąĮ-5-čéčĆąĖč乊čüčäą░čé čåąĖą║ą╗ąŠčģąĖą┤čĆąŠą╗ą░ąĘą░, ąĖčéą┤.). ąÆąĄčøąĖąĮą░ čüą╗čāčćą░čśąĄą▓ą░ čäą░čéą░ą╗ąĮąĖčģ čüą╗čāčćą░čśąĄą▓ą░ čśąĄ ąĘą░ą▒ąĄą╗ąĄąČąĄąĮą░ ą║ąŠą┤ ą┐ą░čåąĖčśąĄąĮą░čéą░ čüą░ čĆąĄčéą║ąĖą╝ ą▓ą░čĆąĖčśą░čåąĖčśą░ą╝ą░ čā ą¤ąÜąŻ, čüą░ ą║ą╗ąĖąĮąĖčćą║ąĖą╝ ą╝ą░ąĮąĖč乥čüčéą░čåąĖčśą░ą╝ą░ čüą▓ąĖčģ ąŠą▒ą╗ąĖą║ą░ ą▒ąŠą╗ąĄčüčéąĖ čüą╗ąĖčćąĮąĖčģ. ąĀąĖąĘąĖą║ ą┐ąŠčĆąŠčÆą░čśą░ čüą░ ą╝čāčéąĖčĆą░ąĮąĖą╝ ą│ąĄąĮąŠą╝ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ čģąĖą┤čĆąŠą║čüąĖą╗ą░ąĘąĄ čüąĄ ą┐ąŠą▓ąĄčøą░ą▓ą░ ą░ą║ąŠ čüčā čĆąŠą┤ąĖč鹥čÖąĖ ą▒ą╗ąĖčüą║ąĖ čĆąŠčÆą░čåąĖ (čüą░ čāčüą║ąŠ ą┐ąŠą▓ąĄąĘą░ąĮąĖą╝ ą▒čĆą░ą║ąŠą▓ąĖą╝ą░).
ąöąĖčśą░ą│ąĮąŠčüčéąĖą║ą░
ąŻ čüą╗čāčćą░čśčā čüčāą╝čÜąĄ ąĮą░ ą│ąĄąĮąĄčéčüą║ąĖ ą┐ąŠčĆąĄą╝ąĄčøą░čś, ą┤ąĖčśą░ą│ąĮąŠąĘą░ čüąĄ čāčéą▓čĆčÆčāčśąĄ ąĮą░ ąŠčüąĮąŠą▓čā ąĘą▒ąĖčĆąĮąĖčģ ą┐ąŠą┤ą░čéą░ą║ą░ ą┤ąŠą▒ąĖčśąĄąĮąĖčģ ą║ą░ąŠ čĆąĄąĘčāą╗čéą░čé ąĖčüčéčĆą░ąČąĖą▓ą░čÜą░ ą░ąĮą░ą╝ąĮąĄąĘąĄ ą▒ąŠą╗ąĄčüčéąĖ - ą│ąĄąĮąĄą░ą╗ąŠčłą║ąĄ ą┐ąŠą┤ą░čéą║ąĄ, čĆąĄąĘčāą╗čéą░č鹥 ą║ą╗ąĖąĮąĖčćą║ąĖčģ ąĖ ą╝ąĄą┤ąĖčåąĖąĮčüą║ąĖčģ ą│ąĄąĮąĄčéąĖčćą║ąĖčģ ąĖčüčéčĆą░ąČąĖą▓ą░čÜą░. ąŚą░ ą┐čĆą░ą▓ąŠą▓čĆąĄą╝ąĄąĮąŠ ąŠčéą║čĆąĖą▓ą░čÜąĄ čāčĆąŠčÆąĄąĮąĖčģ ą▒ąŠą╗ąĄčüčéąĖ (ą¤ąÜąŻ, čåąĖčüčéąĖčćąĮą░ čäąĖą▒čĆąŠąĘą░, ą│ą░ą╗ą░ą║č鹊čüąĄą╝ąĖčśą░, ąĖčéą┤.) ą¤čĆąŠą│čĆą░ą╝ ąŠą▒ą░ą▓ąĄąĘąĮąĄ ą╝ą░čüąĄąøą░ą▒ąŠčĆą░č鹊čĆąĖčśčüą║ąŠ ąĖčüą┐ąĖčéąĖą▓ą░čÜąĄ čüą▓ąĖčģ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ (ąĮąĄąŠąĮą░čéą░ą╗ąĮąĖ čüą║čĆąĖąĮąĖąĮą│).
ąÉą║ąŠ čüčā ą▒čāą┤čāčøąĖ čĆąŠą┤ąĖč鹥čÖąĖ čüą▓ąĄčüąĮąĖ ą┐čĆąĖčüčāčüčéą▓ą░ ą╝čāčéąĖčĆą░ąĮąŠą│ ą│ąĄąĮą░, čüą░ą▓čĆąĄą╝ąĄąĮą░ ą╝ąĄą┤ąĖčåąĖąĮą░ ąĮčāą┤ąĖ ąĮą░čćąĖąĮąĄ ąŠčéą║čĆąĖą▓ą░čÜą░ ą┤ąĄč乥ą║čéą░ č鹊ą║ąŠą╝ čéčĆčāą┤ąĮąŠčøąĄ (ą┐čĆąĄąĮą░čéą░ą╗ąĮą░ ą┤ąĖčśą░ą│ąĮąŠąĘą░ č乥čéčāčüą░ ąĖąĮą▓ą░ąĘąĖą▓ąĮąŠą╝ ą╝ąĄč鹊ą┤ąŠą╝). ąŚą░ ą┐ąŠą┤ąĄą╗čā č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśąĄ ą┐čĆąĄą╝ą░ ą▓čĆčüčéąĖ ąŠąĘą▒ąĖčÖąĮąŠčüčéąĖ, ą║ąŠčĆąĖčüčéąĖ čüąĄ čāčüą╗ąŠą▓ąĮą░ ą║ą╗ą░čüąĖčäąĖą║ą░čåąĖčśą░ ąĮą░ ąŠčüąĮąŠą▓čā ąĮąĖą▓ąŠą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā čäąĖą▒čĆąĖąĮąŠą│ąĄąĮąŠčś č鹥čćąĮąŠčüčéąĖ ą┤ąŠą▒ąĖčśąĄąĮąŠčś ąĖąĘ ą┐ą╗ą░ąĘą╝ą░ ą┐ą╗ą░ąĘą╝ąĄ:
- ąóąĄčłą║ą░ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ - 1200 ą╝ą║ą╝ąŠą╗ /ą╗.
- ą¤čĆąŠčüąĄčćąĮąŠ - 60-1200 ╬╝ą╝ąŠą╗ /ą╗.
- ąøą░ą║ąŠ (ąĮąĖčśąĄ ą┐ąŠčéčĆąĄą▒ą░ąĮ čéčĆąĄčéą╝ą░ąĮ) - 480 ╬╝ą╝ąŠą╗ /ą╗.
ąóąĄčüčé ą┐čĆąŠą▒ąĖčĆą░
ąöąĄč鹥ą║čåąĖčśą░ ą│ąĄąĮąĄčéčüą║ąĖčģ ą░ą▒ąĮąŠčĆą╝ą░ą╗ąĮąŠčüčéąĖ ąŠą┤ą▓ąĖčśą░ čüąĄ čā ąĮąĄą║ąŠą╗ąĖą║ąŠ čäą░ąĘą░. ąŻ ą┐čĆą▓ąŠčś čäą░ąĘąĖ čā ą▒ąŠą╗ąĮąĖčåąĖ ą║ąŠą┤ čüą▓ąĖčģ ą▒ąĄą▒ą░ čüąĄ ą▓čĆčłąĖ 3-5 ą┤ą░ąĮą░ ąČąĖą▓ąŠčéą░, čāąĘąĖą╝ą░čÜąĄ čāąĘąŠčĆą░ą║ą░ ą┐ąĄčĆąĖč乥čĆąĮąĄ ą║čĆą▓ąĖ (ąŠą┤ ą┐ąĄčé) ąĘą░ ąĖčüčéčĆą░ąČąĖą▓ą░čÜąĄ. ą£ą░č鹥čĆąĖčśą░ą╗ čüąĄ ąĮą░ąĮąŠčüąĖ ąĮą░ ą┐ą░ą┐ąĖčĆąĮą░čéąĖ ąŠą▒ą╗ąĖą║ ąĖ čłą░čÖąĄ čā ą▒ąĖąŠčģąĄą╝ąĖčśčüą║čā ą╗ą░ą▒ąŠčĆą░č鹊čĆąĖčśčā, ą│ą┤čśąĄ čüąĄ ąŠą┤ą▓ąĖčśą░ ą▒ąĖąŠą║ąĄą╝ąĖčśčüą║ą░ ą░ąĮą░ą╗ąĖąĘą░. ąŻ ą┤čĆčāą│ąŠčś čäą░ąĘąĖ čüą║čĆąĖąĮąĖąĮą│ č鹥čüčéą░, ąŠą┤čĆąĄčÆąĄąĮą░ čśąĄ ą║ąŠąĮčåąĄąĮčéčĆą░čåąĖčśą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ ąĮąŠčĆą╝ą░ą╗ąĮąĄ ą▓čĆąĖčśąĄą┤ąĮąŠčüčéąĖ.

ąÉą║ąŠ čüąĄ ąĮąĄ ąŠčéą║čĆąĖčśčā ą┐ą░č鹊ą╗ąŠčłą║ąĄ ą┐čĆąŠą╝čśąĄąĮąĄ, ą┤ąĖčśą░ą│ąĮąŠąĘą░ čüąĄ ąĘą░ą▓čĆčłą░ą▓ą░, čłč鹊 čüąĄ ą▒ąĖčÖąĄąČąĖ čā ąĖčüą║ą░ąĘąĮąĖčåąĖ ą┤čśąĄč鹥čéą░. ąŻ ą┐čĆąĖčüčāčüčéą▓čā ąŠą┤čüčéčāą┐ą░čÜą░ ąŠą┤ ąĮąŠčĆą╝ąĄ, čĆąĄąĘčāą╗čéą░čéąĖ ą┤ąĖčśą░ą│ąĮąŠąĘąĄ čüąĄ čłą░čÖčā ą╗ąĄą║ą░čĆčā-ą┐ąĄą┤ąĖčśą░čéčĆčā čĆą░ą┤ąĖ ą┤ąŠčĆą░ą┤ąĄ čüčéčāą┤ąĖčśąĄ čāąĘąŠčĆą║ą░ ą║čĆą▓ąĖ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮč湥čéą░. ąŚą┤čĆą░ą▓čÖąĄ ą┤ąĄč鹥čéą░ ąĘą░ą▓ąĖčüąĖ ąŠą┤ ą▒ą╗ą░ą│ąŠą▓čĆąĄą╝ąĄąĮąŠą│ ąĖ čéą░čćąĮąŠą│ ąĖąĘą▓čĆčłą░ą▓ą░čÜą░ čüą▓ąĖčģ ą╝ąĄčĆą░ ąĘą░ ąŠčéą║čĆąĖą▓ą░čÜąĄ ą░ą▒ąĮąŠčĆą╝ą░ą╗ąĮąŠčüčéąĖ. ąÉą║ąŠ čüąĄ ą┤ąĖčśą░ą│ąĮąŠąĘą░ ą┐ąŠčéą▓čĆą┤ąĖ ąĮą░ą║ąŠąĮ ą┐ąŠąĮąŠą▓čÖąĄąĮąŠą│ č鹥čüčéą░, čĆąŠą┤ąĖč鹥čÖąĖ ą┤ąĄč鹥čéą░ čģąŠčøąĄą┐ąŠčüą╗ą░čéąĖ čā ą║ą╗ąĖąĮąĖą║čā ąĘą░ ą┤čśąĄčćčśčā ą│ąĄąĮąĄčéąĖą║čā čĆą░ą┤ąĖ ą╗ąĖčśąĄč湥čÜą░.
ąÉąĮą░ą╗ąĖąĘąĄ ąĖ čüčéčāą┤ąĖčśąĄ ąĘą░ ą┐ąŠčéą▓čĆą┤čā ą┤ąĖčśą░ą│ąĮąŠąĘąĄ
ą¤ąŠąĮąŠą▓čÖąĄąĮą░ ą┤ąĖčśą░ą│ąĮąŠąĘą░ čā čüą╗čāčćą░čśčā ąŠčéą║čĆąĖą▓ą░čÜą░ ąŠą┤čüčéčāą┐ą░čÜą░ ąŠą┤ ąĮąŠčĆą╝ąĄ č鹊ą║ąŠą╝ ą┐ąŠč湥čéąĮąŠą│ ą┐čĆąŠą▒ąĖčĆąĮąŠą│ č鹥čüčéą░ ą▓čĆčłąĖ čüąĄ ą┐ąŠąĮąŠą▓ąĮąŠą╝ ą┐čĆąŠčåčśąĄąĮąŠą╝ ą░ąĮą░ą╗ąĖąĘą░. ą¤ąŠčĆąĄą┤ ąŠą┤čĆąĄčÆąĖą▓ą░čÜą░ čüą░ą┤čƹȹ░čśą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā ą║čĆą▓ąĖ ąĮą░ ą╝ąĄč鹊ą┤ąĄ ą┤ąĖčśą░ą│ąĮąŠąĘąĄ ą¤ąÜąŻ ą║ąŠą┤ ą┤ąĄčåąĄ ąĖ ąŠą┤čĆą░čüą╗ąĖčģ čāą║čÖčāčćčāčśčā:
- ąśčüą┐ąĖčéąĖą▓ą░čÜąĄ čüąĄč湥čÜą░ - ąŠą┤čĆąĄčÆąĖą▓ą░čÜąĄ č乥ąĮąĖą╗ą┐ąĖčĆčāą▓ąĖčćąĮąĄ ą║ąĖčüąĄą╗ąĖąĮąĄ čā čāčĆąĖąĮčā ą┤ąŠą┤ą░ą▓ą░čÜąĄą╝ ą│ą▓ąŠąČčÆą░ čā ą▒ąĖąŠą╝ą░č鹥čĆąĖčśą░ą╗ (ą┐ąŠčüč鹊蜹Ė ą┐ą╗ą░ą▓ąŠ-ąĘąĄą╗ąĄąĮą░ ą▒ąŠčśą░);
- ąōą░čéčĆąĄąĄ č鹥čüčé - ą┐čĆąŠčåąĄąĮą░ čüč鹥ą┐ąĄąĮą░ čĆąĄą░ą║čåąĖčśąĄ ą╝ąĖą║čĆąŠąŠčĆą│ą░ąĮąĖąĘą░ą╝ą░ ąĮą░ ą╝ąĄčéą░ą▒ąŠą╗ąĖčćą║ąĄ ą┐čĆąŠą┤čāą║č鹥 ąĖą╗ąĖ ąĄąĮąĘąĖą╝ąĄ ą║ąŠčśąĖ čüąĄ ąĮą░ą╗ą░ąĘąĄ čā ą║čĆą▓ąĖ ą┐ą░čåąĖčśąĄąĮčéą░;
- čģčĆąŠą╝ą░č鹊ą│čĆą░čäąĖčśą░ - ą┐čĆąŠčāčćą░ą▓ą░čÜąĄ čģąĄą╝ąĖčśčüą║ąĖčģ čüą▓ąŠčśčüčéą░ą▓ą░ čüčāą┐čüčéą░ąĮčåąĖ čĆą░čüą┐ąŠčĆąĄčÆąĄąĮąĖčģ ąĖąĘą╝ąĄčÆčā ą┤ą▓ąĄ čäą░ąĘąĄ;
- čäą╗čāąŠčĆąĖą╝ąĄčéčĆąĖčśą░ - ąĘčĆą░č湥čÜąĄ ą▒ąĖąŠą╝ą░č鹥čĆąĖčśą░ą╗ą░ ą╝ąŠąĮąŠčģčĆąŠą╝ą░čéčüą║ąĖą╝ ąĘčĆą░č湥čÜąĄą╝ ąĘą░ ąŠą┤čĆąĄčÆąĖą▓ą░čÜąĄ ą║ąŠąĮčåąĄąĮčéčĆą░čåąĖčśąĄ čüčāą┐čüčéą░ąĮčåąĖ ą║ąŠčśąĄ čüąĄ čā čÜąŠčś ąĮą░ą╗ą░ąĘąĄ;
- ąĄą╗ąĄą║čéčĆąŠąĄąĮčåąĄčäą░ą╗ąŠą│čĆą░čäąĖčśą░ - ą┤ąĖčśą░ą│ąĮąŠčüčéąĖą║ą░ ąĄą╗ąĄą║čéčĆąĖčćąĮąĄ ą░ą║čéąĖą▓ąĮąŠčüčéąĖ ą╝ąŠąĘą│ą░;
- čüąĮąĖą╝ą░čÜąĄ ą╝ą░ą│ąĮąĄčéąĮąŠą╝ čĆąĄąĘąŠąĮą░ąĮčåąĖčśąŠą╝ - ą║čĆčłąĄčÜąĄ ą░č鹊ą╝čüą║ąĖčģ čśąĄąĘą│čĆą░ čøąĄą╗ąĖčśą░ ąĄą╗ąĄą║čéčĆąŠą╝ą░ą│ąĮąĄčéąĮąĖą╝ čéą░ą╗ą░čüąĖą╝ą░ ąĖ ą╝ąĄčĆąĄčÜąĄ čÜąĖčģąŠą▓ąŠą│ ąŠą┤ą│ąŠą▓ąŠčĆą░.
ąøąĄč湥čÜąĄ ą║ą╗ą░čüąĖčćąĮąĄ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖąĄ
ą×čüąĮąŠą▓ą░ č鹥čĆą░ą┐ąĖčśąĄ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čśąĄ ąŠą│čĆą░ąĮąĖč湥čÜąĄ ą┐ąŠčéčĆąŠčłčÜąĄ ą┐čĆąŠąĖąĘą▓ąŠą┤ą░, ą║ąŠčśąĖ čśąĄ ąĖąĘą▓ąŠčĆ ą┐čĆąŠč鹥ąĖąĮą░ ąČąĖą▓ąŠčéąĖčÜčüą║ąŠą│ ąĖ ą▒ąĖčÖąĮąŠą│ ą┐ąŠčĆąĖčśąĄą║ą╗ą░. ąłąĄą┤ąĖąĮąĖ ąĮą░čćąĖąĮ čāčüą┐ąĄčłąĮąŠą│ ą╗ąĄč湥čÜą░ čśąĄ ą┤ąĖčśąĄčéą░ą╗ąĮą░ č鹥čĆą░ą┐ąĖčśą░, čćąĖčśą░ čśąĄ ą░ą┤ąĄą║ą▓ą░čéąĮąŠčüčé ą┐čĆąŠčåąĄčÜąĄąĮą░ čüą░ą┤čƹȹ░čśąĄą╝ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā čüąĄčĆčāą╝čā. ą£ą░ą║čüąĖą╝ą░ą╗ąĮąŠ ą┤ąŠąĘą▓ąŠčÖąĄąĮąĖ ąĮąĖą▓ąŠ ą░ą╝ąĖąĮąŠą║ąĖčüąĄą╗ąĖąĮą░ ą║ąŠą┤ ą┐ą░čåąĖčśąĄąĮą░čéą░ čĆą░ąĘą╗ąĖčćąĖčéąĖčģ čüčéą░čĆąŠčüąĮąĖčģ ą│čĆčāą┐ą░čśąĄ:
- ą║ąŠą┤ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮčćą░ą┤ąĖ ąĖ ą┤čśąĄčåąĄ ą╝ą╗ą░čÆąĄ ąŠą┤ 3 ą│ąŠą┤ąĖąĮąĄ - ą┤ąŠ 242 ╬╝ą╝ąŠą╗ /ą╗;
- ąĘą░ ą┐čĆąĄą┤čłą║ąŠą╗čåąĄ - ą┤ąŠ 360 ą╝ą║ą╝ąŠą╗ /ą╗;
- ą║ąŠą┤ ą┐ą░čåąĖčśąĄąĮą░čéą░ ąŠą┤ 7 ą┤ąŠ 14 ą│ąŠą┤ąĖąĮą░ - ą┤ąŠ 480 ╬╝ą╝ąŠą╗ /ąø;
- ą║ąŠą┤ ą░ą┤ąŠą╗ąĄčüčåąĄąĮą░čéą░ - ą┤ąŠ 600 ╬╝ą╝ąŠą╗ /ą╗.
ąĢč乥ą║ą░čé ąĖčüčģčĆą░ąĮąĄ ąĘą░ą▓ąĖčüąĖ ąŠą┤ č鹊ą│ą░ čā ą║ąŠčśąŠčś čäą░ąĘąĖ ą▒ąŠą╗ąĄčüčéąĖ čśąĄ ą║ąŠčĆąĄą║čåąĖčśą░ ąĖčüčģčĆą░ąĮąĄ. ąÜąŠą┤ čĆą░ąĮąĄ ą┤ąĖčśą░ą│ąĮąŠąĘąĄ ą║ąŠąĮą│ąĄąĮąĖčéą░ą╗ąĮąĄ ą┐ą░č鹊ą╗ąŠą│ąĖčśąĄ ą┐čĆąŠą┐ąĖčüą░ąĮą░ čśąĄ ą┤ąĖčśąĄčéą░ą╗ąĮą░ č鹥čĆą░ą┐ąĖčśą░ čā čéčĆą░čśą░čÜčā ąŠą┤ 8 ąĮąĄą┤ąĄčÖą░ ąČąĖą▓ąŠčéą░ (ąĮą░ą║ąŠąĮ ąŠą▓ąŠą│ ą┐ąĄčĆąĖąŠą┤ą░ ą▓ąĄčø čüąĄ ą┐ąŠčćąĖčÜčā ąĮąĄą┐ąŠą▓čĆą░čéąĮąĄ ą┐čĆąŠą╝ąĄąĮąĄ). ąØąĄą┤ąŠčüčéą░čéą░ą║ ą▒ą╗ą░ą│ąŠą▓čĆąĄą╝ąĄąĮąĖčģ ą╝ąĄčĆą░ ą┤ąŠą▓ąŠą┤ąĖ ą┤ąŠ ą║ąŠą╝ą┐ą╗ąĖą║ą░čåąĖčśą░ ąĖ čüą╝ą░čÜąĄčÜą░ ąĮąĖą▓ąŠą░ ąĖąĮč鹥ą╗ąĖą│ąĄąĮčåąĖčśąĄ ąĘą░ 4 ą▒ąŠą┤ą░ čā čśąĄą┤ąĮąŠą╝ ą╝ąĄčüąĄčåčā ąŠą┤ čĆąŠčÆąĄčÜą░ ą┤ąŠ ą┐ąŠč湥čéą║ą░ ą╗ąĄč湥čÜą░.

ąĪ ąŠą▒ąĘąĖčĆąŠą╝ ąĮą░ č鹊 ą┤ą░ č鹥čĆą░ą┐ąĖčśčüą║ą░ ą┤ąĖčśąĄčéą░ ąĘą░ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśčā čāą║čÖčāčćčāčśąĄ ą┐ąŠčéą┐čāąĮąŠ ąĖčüą║čÖčāčćąĖą▓ą░čÜąĄ ąĖąĘ ą┐čĆąĄčģčĆą░ąĮąĄ ąČąĖą▓ąŠčéąĖčÜčüą║ąĖčģ ą┐čĆąŠč鹥ąĖąĮą░, ąĮąĄąŠą┐čģąŠą┤ąĮąŠ čśąĄ ą║ąŠčĆąĖčüčéąĖčéąĖ ą┤čĆčāą│ąĄ ąĖąĘą▓ąŠčĆąĄ ąĄčüąĄąĮčåąĖčśą░ą╗ąĮąĖčģ ą░ą╝ąĖąĮąŠą║ąĖčüąĄą╗ąĖąĮą░, ą║ą░ąŠ ąĖ ą▓ąĖčéą░ą╝ąĖąĮąĄ ą│čĆčāą┐ąĄ ąæ, ą╝ąĖąĮąĄčĆą░ą╗ąĮąĖčģ čśąĄą┤ąĖčÜąĄčÜą░ ą║ąŠčśą░ čüą░ą┤čƹȹĄ ą║ą░ą╗čåąĖčś ąĖ č乊čüč乊čĆ. ą¤čĆąŠąĖąĘą▓ąŠą┤ąĖ ą║ąŠčśąĖ čüčā ąĮą░ą╝ąĄčÜąĄąĮąĖ ąĘą░ ą┤ąŠą┤ą░čéą░ą║ ą┐čĆąĄčģčĆą░ąĮąĖ ą▒ąĄąĘ ą┐čĆąŠč鹥ąĖąĮą░ čāą║čÖčāčćčāčśčā:
- ą┐čĆąŠč鹥ąĖąĮ čģąĖą┤čĆąŠą╗ąĖąĘą░čéąĖ (ą░ą╝ąĖą│ąĄąĮąĖ, ą░ą╝ąĖąĮą░ąĘąŠą╗, čäąĖą▒čĆąĖąĮąŠąĘąĄ);
- ąĮąĄ čüą░ą┤čƹȹĄ čüą╝ąĄčłąĄ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ ąĘą░čüąĖčøąĄąĮąĄ ąĄčüąĄąĮčåąĖčśą░ą╗ąĮąĖą╝ ą░ą╝ąĖąĮąŠą║ąĖčüąĄą╗ąĖąĮą░ą╝ą░ - ąóąĄčéčĆą░č乥ąĮ, č乥ąĮąĖą╗ čüą╗ąŠą▒ąŠą┤ą░ąĮ.
ąŚą░čśąĄą┤ąĮąŠ čüą░ ą║čāčĆą░čéąĖą▓ąĮąĖą╝ ą╝čśąĄčĆą░ą╝ą░ ąĘą░ ąŠčéą║ą╗ą░čÜą░čÜąĄ čāąĘčĆąŠą║ą░ čäčāąĮą║čåąĖąŠąĮąĖčüą░čÜą░ ąŠčĆą│ą░ąĮąĖąĘą╝ą░, ą┐ąŠčéčĆąĄą▒ąĮąŠ čśąĄ ąĖąĘą▓čĆčłąĖčéąĖ čüąĖą╝ą┐č鹊ą╝ą░čéčüą║ąŠ ą╗ąĖčśąĄč湥čÜąĄ čāčüą╝čśąĄčĆąĄąĮąŠ ąĮą░ ąŠčéą║ą╗ą░čÜą░čÜąĄ ą┤ąĄč乥ą║ą░čéą░ ą│ąŠą▓ąŠčĆą░ ąĖ ąĮąŠčĆą╝ą░ą╗ąĖąĘą░čåąĖčśčā ą║ąŠąŠčĆą┤ąĖąĮą░čåąĖčśąĄ ą┐ąŠą║čĆąĄčéą░. ąÜąŠą╝ą┐ą╗ąĄą║čüąĮą░ č鹥čĆą░ą┐ąĖčśą░ čāą║čÖčāčćčāčśąĄ čäąĖąĘąĖąŠč鹥čĆą░ą┐ąĖčśčüą║ąĄ ą┐čĆąŠčåąĄą┤čāčĆąĄ, ą╝ą░čüą░ąČčā, ą┐ąŠą╝ąŠčø ą╗ąŠą│ąŠą┐ąĄą┤ą░, ą┐čüąĖčģąŠą╗ąŠą│ą░, ąĖąĘą▓ąŠčÆąĄčÜąĄ ą│ąĖą╝ąĮą░čüčéąĖčćą║ąĖčģ ą▓čśąĄąČą▒ąĖ. ąŻ ąĮąĄą║ąĖą╝ čüą╗čāčćą░čśąĄą▓ąĖą╝ą░, čāąĘ ą┤ąĖčśąĄčéą░ą╗ąĮčā č鹥čĆą░ą┐ąĖčśčāą┐ąŠą║ą░ąĘčāčśąĄ čāą┐ąŠčéčĆąĄą▒čā ą░ąĮčéąĖą║ąŠąĮą▓čāą╗ąĘąĖą▓ą░, ąĮąŠąŠčéčĆąŠą┐ąĮąĖčģ ąĖ ą▓ą░čüą║čāą╗ą░čĆąĮąĖčģ ą╗ąĄą║ąŠą▓ą░.
ąÜą░čĆą░ą║č鹥čĆąĖčüčéąĖą║ąĄ čéčĆąĄčéą╝ą░ąĮą░ ą░čéąĖą┐ąĖčćąĮąĖčģ ąŠą▒ą╗ąĖą║ą░
ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čéąĖą┐ą░ ąśąś ąĖ ąśąśąś ąĮąĄ ą╝ąŠąČąĄ čüąĄ ą╗ąĄčćąĖčéąĖ ąĖčüčģčĆą░ąĮąŠą╝ čüą░ ąĮąĖčüą║ąĖą╝ čüą░ą┤čƹȹ░čśąĄą╝ ą┐čĆąŠč鹥ąĖąĮą░ - ąĮąĖą▓ąŠ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā ą║čĆą▓ąĖ ąŠčüčéą░čśąĄ ąĮąĄą┐čĆąŠą╝ąĄčÜąĄąĮ čā ąŠą│čĆą░ąĮąĖčćą░ą▓ą░čÜčā ą┐čĆąŠč鹊ą║ą░ ą┐čĆąŠč鹥ąĖąĮą░ čā ąŠčĆą│ą░ąĮąĖąĘą╝čā, ąĖą╗ąĖ ą║ą╗ąĖąĮąĖčćą║ąĖ čüąĖą╝ą┐č鹊ą╝ąĖ ąĮą░ą┐čĆąĄą┤čāčśčā čćą░ą║ ąĖ ą║ą░ą┤ą░ čüąĄ ąĮąĖą▓ąŠ ą░ą╝ąĖąĮąŠą║ąĖčüąĄą╗ąĖąĮąĄ čüą╝ą░čÜąĖ. ąĢč乥ą║čéąĖą▓ąĮą░ č鹥čĆą░ą┐ąĖčśą░ ąŠą▓ąĖčģ ąŠą▒ą╗ąĖą║ą░ ą▒ąŠą╗ąĄčüčéąĖ ąŠą┤ą▓ąĖčśą░ čüąĄ čāąĘ ą┐čĆąĖą╝ąĄąĮčā:
- č鹥čéčĆą░čģąĖą┤čĆąŠą▒ąĖąŠą┐č鹥čĆąĖąĮ - čäą░ą║č鹊čĆ čāčéąĖčåą░čśą░ ąĄąĮąĘąĖą╝ą░;
- čüąĖąĮč鹥čéčüą║ąĖ ą░ąĮą░ą╗ąŠąĘąĖ č鹥čéčĆą░čģąĖą┤čĆąŠą▒ąĖąŠą┐č鹥čĆąĖąĮą░ - č鹥 čüčāą┐čüčéą░ąĮčåąĄ ą▒ąŠčÖąĄ ą┐čĆąŠą┤ąĖčĆčā ą║čĆąŠąĘ ą║čĆą▓ąĮąŠ-ą╝ąŠąČą┤ą░ąĮčā ą▒ą░čĆąĖčśąĄčĆčā;
- ą╗ąĖčśąĄą║ąŠą▓ąĖ ąĘą░ čüčāą┐čüčéąĖčéčāčåąĖčśčüą║čā č鹥čĆą░ą┐ąĖčśčā - ąĮąĄ čāą║ą╗ą░čÜą░čśčā čāąĘčĆąŠą║ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśąĄ, ą▓ąĄčø ą┐ąŠą┤čƹȹ░ą▓ą░čśčā ąĮąŠčĆą╝ą░ą╗ąĮąŠ čäčāąĮą║čåąĖąŠąĮąĖčĆą░čÜąĄ čéąĖčśąĄą╗ą░ (ą╗ąĄą▓ąŠą┤ąŠą┐ą░ čā ą║ąŠą╝ą▒ąĖąĮą░čåąĖčśąĖ čü ą║ą░čĆą▒ąĖą┤ąŠą┐ąŠą╝, 5-ąŠą║čüąĖčéčĆąĖą┐č鹊čäą░ąĮąŠą╝, 5-č乊čĆą╝ąĖą╗č鹥čéčĆą░čģąĖą┤čĆąŠč乊ą╗ą░č鹊ą╝);
- čģąĄą┐ą░č鹊ą┐čĆąŠč鹥ą║č鹊čĆąĖ - ą┐ąŠą┤čƹȹ░ą▓ą░čśčā čäčāąĮą║čåąĖąŠąĮąĖčüą░čÜąĄ čśąĄčéčĆąĄ;
- ą░ąĮčéąĖą║ąŠąĮą▓čāą╗ąĘąĖą▓ąĖ;
- čāą▓ąŠčÆąĄčÜąĄ ą│ąĄąĮą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ čģąĖą┤čĆąŠą║čüąĖą╗ą░ąĘąĄ čā čśąĄčéčĆčā - ąĄą║čüą┐ąĄčĆąĖą╝ąĄąĮčéą░ą╗ąĮą░ ą╝ąĄč鹊ą┤ą░.
ąÜą░čĆą░ą║č鹥čĆąĖčüčéąĖą║ąĄ ąĖčüčģčĆą░ąĮąĄ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮč湥čéą░ ąĖ č鹥čĆą░ą┐ąĖčśą░ ąĖčüčģčĆą░ąĮąĄ
ąŻ ą┐čĆą▓ąŠčś ą│ąŠą┤ąĖąĮąĖ ąČąĖą▓ąŠčéą░ ą┤čśąĄč鹥čéą░, ą┤ąŠą┐čāčłč鹥ąĮąŠ čśąĄ ą┤ąŠčśąĄčÜąĄ, ą░ą╗ąĖ čÜąĄą│ąŠą▓ą░ ą║ąŠą╗ąĖčćąĖąĮą░ čéčĆąĄą▒ą░ ą▒ąĖčéąĖ ąŠą│čĆą░ąĮąĖč湥ąĮą░. ąöąŠ 6 ą╝ąĄčüąĄčåąĖ, ą┐čĆąĖčģą▓ą░čéčÖąĖą▓ąĖ ąĮąĖą▓ąŠ ą┐ąŠčéčĆąŠčłčÜąĄ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čśąĄ 60-90 ą╝ą│ ąĮą░ 1 ą║ą│ č鹥ąČąĖąĮąĄ ą▒ąĄą▒ąĄ (čā 100 ą│ ą╝ą╗ąĄą║ą░ čüą░ą┤čƹȹĖ 5.6 ą╝ą│ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░). ą¤ąŠč湥ą▓čłąĖ ąŠą┤ 3 ą╝ąĄčüąĄčåą░, ą▒ąĄą▒ąĖąĮą░ ąĖčüčģčĆą░ąĮą░ čéčĆąĄą▒ą░ ą┐ąŠčüč鹥ą┐ąĄąĮąŠ ą┐ąŠą▓ąĄčøą░ą▓ą░čéąĖ čāą▓ąŠčÆąĄčÜąĄą╝ ą▓ąŠčøąĮąĖčģ čüąŠą║ąŠą▓ą░ ąĖ ą┐ąĖčĆąĄą░.
ąŚą░ ą┤ąĄčåčā ąŠą┤ 6 ą╝ąĄčüąĄčåąĖ ą┤ąŠąĘą▓ąŠčÖąĄąĮąŠ čśąĄ čāąĮąŠčłąĄčÜąĄ čā ąĖčüčģčĆą░ąĮčā ą▒ąĖčÖąĮąŠą│ ą┐ąĖčĆąĄą░, ą║ą░čłąĄ (ąĖąĘ čüą░ą│ą░), ą▒ąĄąĘ ą┐čĆąŠč鹥ąĖąĮą░ą║ąĖčüčüąĄčü ą¤ąŠčüą╗ąĄ 7 ą╝ąĄčüąĄčåąĖ ą▒ąĄą▒ąĖ ą╝ąŠąČąĄč鹥 ą┤ą░čéąĖ č鹥čüč鹥ąĮąĖąĮčā čüą░ ąĮąĖčüą║ąĖą╝ ą┐čĆąŠč鹥ąĖąĮąŠą╝, čüą░ 8 ą╝ąĄčüąĄčåąĖ - čģą╗ąĄą▒ąŠą╝ ą▒ąĄąĘ ą┐čĆąŠč鹥ąĖąĮą░. ąØąĖčśąĄ čāčéą▓čĆčÆąĄąĮąŠ čüčéą░čĆąŠčüąĮąŠ ą┤ąŠą▒ą░ čā ą║ąŠą╝ąĄ čéčĆąĄą▒ą░ ąŠą│čĆą░ąĮąĖčćąĖčéąĖ ą┐čĆąĄąĮąŠčü ą┐čĆąŠč鹥ąĖąĮą░ čā ąŠčĆą│ą░ąĮąĖąĘą╝čā ą▒ąŠą╗ąĄčüąĮąŠą│ ą┤čśąĄč鹥čéą░. ąøąĄą║ą░čĆąĖ ąĖ ą┤ą░čÖąĄ čĆą░ąĘą╝ą░čéčĆą░čśčā ą┐ąĖčéą░čÜąĄ ąĄčäąĖą║ą░čüąĮąŠčüčéąĖ ą┤ąĖčśąĄč鹥čéčüą║ąĄ č鹥čĆą░ą┐ąĖčśąĄ, ą░ą╗ąĖ čüąĄ čüą╗ą░ąČčā ą┤ą░ ąĮą░čśą╝ą░čÜąĄ 18 ą│ąŠą┤ąĖąĮą░, ą┐čĆąĄčģčĆą░ą╝ą▒ąĄąĮą░ ąĖčüčģčĆą░ąĮą░ čéčĆąĄą▒ą░ ą┐ąŠčłč鹊ą▓ą░čéąĖ.
ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░, ą┤ąĖčśą░ą│ąĮąŠčüčéąĖą║ąŠą▓ą░ąĮą░ čüą░ ąČąĄąĮąŠą╝, ąĮąĖčśąĄ čĆą░ąĘą╗ąŠą│ ąĘą░ ąĮą░ą┐čāčłčéą░čÜąĄ čĆąŠčÆąĄčÜą░ ą┤čśąĄč鹥čéą░. ąæčāą┤čāčøąĄ ą╝ą░čśą║ąĄ čüą░ ą¤ąÜąŻ ąĘą░ čüą┐čĆąĄčćą░ą▓ą░čÜąĄ ąŠčłč鹥čøąĄčÜą░ č乥čéčāčüą░ č鹊ą║ąŠą╝ čéčĆčāą┤ąĮąŠčøąĄ ąĖ ąĘą░ čüą┐čĆąĄčćą░ą▓ą░čÜąĄ ą╝ąŠą│čāčøąĖčģ ą║ąŠą╝ą┐ą╗ąĖą║ą░čåąĖčśą░ ąĮąĄąŠą┐čģąŠą┤ąĮąĄ čüčā ą┐čĆąĄ ą┐ą╗ą░ąĮąĖčĆą░ąĮąŠą│ ąĘą░č湥čøą░ ąĖ č鹊ą║ąŠą╝ ąĮąŠčłąĄčÜą░ ą┤ąĄč鹥čéą░ ą┤ą░ čüąĄ ą┐čĆąĖą┤čƹȹ░ą▓ą░čśčā ąĖčüčģčĆą░ąĮąĄ čāąĘ ąŠą│čĆą░ąĮąĖč湥čÜąĄ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ (ąĮąĖą▓ąŠ čā ą║čĆą▓ąĖ čéčĆąĄą▒ą░ ą┤ą░ ą▒čāą┤ąĄ ą┤ąŠ 242 ą╝ąĖą║čĆąŠą╝ąŠą╗ /ą╗).
ą£ąĄčłą░ą▓ąĖąĮąĄ ą▓ą░ą║čåąĖąĮą░ ąĘą░ ą▒ąĄą▒ąĄ
ąóąŠą╗ąĄčĆą░ąĮčåąĖčśą░ ąĮąŠą▓ąŠčĆąŠčÆąĄąĮč湥čéą░ ą┐čĆąĄą╝ą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮčā čā ą┐čĆą▓ąŠčś ą│ąŠą┤ąĖąĮąĖ ąČąĖą▓ąŠčéą░ ą▒čƹʹŠ čüąĄ ą╝ąĖčśąĄčÜą░, ą┐ą░ čśąĄ ąĮąĄąŠą┐čģąŠą┤ąĮąŠ ą║ąŠąĮčéčĆąŠą╗ąĖčĆą░čéąĖ čÜąĄą│ąŠą▓čā ą║ąŠąĮčåąĄąĮčéčĆą░čåąĖčśčā čā ą║čĆą▓ąĖ ą┤čśąĄč鹥čéą░ ąĖ ą┐čĆąĖą╗ą░ą│ąŠą┤ąĖčéąĖ ąĖčüčģčĆą░ąĮčā. ą£ąĄčłą░ą▓ąĖąĮąĄ čüčā ąĘą░ ąŠą┤čĆąĄčÆąĄąĮąĄ čüčéą░čĆąŠčüąĮąĄ ą│čĆčāą┐ąĄ:
- ąĘą░ ą▒ąĄą▒ąĄ ą┤ąŠ ą│ąŠą┤ąĖąĮą░čéą░ ąĖą╝ąĄąĮąŠą▓ą░ąĮ ąÉčäč乥ąĮąĖą╗ą░ą║ 15, ąÉąĮą░ą╗ąŠą│-ąłąÆ, ą¤ąÜąŻ-1, ą¤ąÜąŻ-ą╝ąĖą║, ą¤ąÜąŻ ąÉąĮą░ą╝ąĖą║;
- ąĘą░ ą┤čśąĄčåčā čüčéą░čĆąĖčśčā ąŠą┤ 1 ą│ąŠą┤ąĖąĮąĄą│ąŠą┤ąĖąĮąĄ, ą┐čĆąŠą┐ąĖčüčāčśčā ąŠą▒ąŠą│ą░čøąĄąĮąĄ ą▓ąĖčéą░ą╝ąĖąĮąĖą╝ą░ ąĖ ą╝ąĖąĮąĄčĆą░ą╗ąĖą╝ą░ ą╝ąĄčłą░ą▓ąĖąĮčā ą▓ąĖčüąŠą║ąŠą│ čüą░ą┤čƹȹ░čśą░ ą┐čĆąŠč鹥ąĖąĮą░ - ą¤ąÜąŻ ą¤čĆąĖą╝ą░, ą¤-ąÉą£ ąŻąĮąĖą▓ąĄčĆčüą░ą╗, ą¤ąÜąŻ-1, ą¤ąÜąŻ-2, ąźąĀą£ ą£ą░ą║ą░ą╝ąĄą┤, ąźąĀ ą£ą░ą║čāą╝čāą╝.

ą¤čĆąĄčģčĆą░ą╝ą▒ąĄąĮąĖ ą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ ąĘą░ ą┤ąŠą┐čāąĮčā ąĘą░ą╗ąĖčģą░ ą┐čĆąŠč鹥ąĖąĮą░
ąłąĄą┤ąĮą░ ąŠą┤ ą│ą╗ą░ą▓ąĮąĖčģ ą║ąŠą╝ą┐ąŠąĮąĄąĮčéąĖ ą┤ąĖčśąĄčéą░ą╗ąĮąĄ ąĖčüčģčĆą░ąĮąĄ ąĘą░ č乥ąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśčā čśąĄ čģčĆą░ąĮą░ čüą░ ą╝ą░ą╗ąŠ ą┐čĆąŠč鹥ąĖąĮą░ ąĮą░ ą▒ą░ąĘąĖ čüą║čĆąŠą▒ą░. ą×ą▓ąĖ ą┤ąŠą┤ą░čåąĖ čüą░ą┤čƹȹĄ čģąĖą┤čĆąŠą╗ąĖąĘą░čé ą║ą░ąĘąĄąĖąĮ, čéčĆąĖą┐č鹊čäą░ąĮ, čéąĖčĆąŠąĘąĖąĮ, ą╝ąĄčéąĖąŠąĮąĖąĮ, ą░ąĘąŠčé ąĖ ąŠą▒ąĄąĘą▒ąĄčÆčāčśčā ą┤ąĮąĄą▓ąĮčā ą┐ąŠčéčĆąĄą▒čā ą┤ąĄč鹥čéą░ ąĘą░ ą┤ąĄčćčśąĖą╝ ą┐čĆąŠč鹥ąĖąĮąŠą╝, ą║ąŠčśąĖ čśąĄ ąĮąĄąŠą┐čģąŠą┤ą░ąĮ ąĘą░ ąĮąŠčĆą╝ą░ą╗ą░ąĮ čĆą░ąĘą▓ąŠčś ąĖ čĆą░čüčé. ąĪą┐ąĄčåąĖčśą░ą╗ąĖąĘąŠą▓ą░ąĮąĖ ą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ, ą║ąŠčśąĖ čā ąĮąĄą┤ąŠčüčéą░čéą║čā ąĖčüčģčĆą░ąĮąĄ ą┐ąŠą┐čāčÜą░ą▓ą░čśčā ąĮąĄą┤ąŠčüčéą░čéą░ą║ ąĮąĄąŠą┐čģąŠą┤ąĮąĖčģ ą╝ąĖąĮąĄčĆą░ą╗ą░ ąĖ ą░ą╝ąĖąĮąŠą║ąĖčüąĄą╗ąĖąĮą░, čüčā:
- ąæąĄčĆą╗ąŠč乥ąĮ;
- ąóčüąĖą╝ąŠčĆą│ą░ąĮ;
- ą£ąĖąĮą░č乥ąĮ; ąÉą┐ąŠč鹊ąĮąĖ.
ą¤čĆąĄčģčĆą░ąĮą░ ąĘą░ ą┐čĆąĄą┤čłą║ąŠą╗čüą║čā ąĖ čłą║ąŠą╗čüą║čā ą┤ąĄčåčā
ąÜą░ą║ąŠ čüąĄ č鹥ą╗ąŠ ą┐čĆąĖą╗ą░ą│ąŠčÆą░ą▓ą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮčā ą║ąŠą┤ ą┤ąĄčåąĄ ąŠą┤ 5 ą│ąŠą┤ąĖąĮą░ čüčéą░čĆąŠčüčéąĖ, ą╝ąŠą│čāčøąĄ čśąĄ ą┐ąŠčüč鹥ą┐ąĄąĮąŠ čüą╝ą░čÜąĖą▓ą░čéąĖ ąŠą│čĆą░ąĮąĖč湥čÜą░ čā ąĖčüčģčĆą░ąĮąĖ. ą¤čĆąŠčłąĖčĆąĄčÜąĄ ąĖčüčģčĆą░ąĮąĄ čśąĄ ą║čĆąŠąĘ čāą▓ąŠčÆąĄčÜąĄ ąČąĖčéą░čĆąĖčåą░, ą╝ą╗ąĄčćąĮąĖčģ ą┐čĆąŠąĖąĘą▓ąŠą┤ą░, ą╝ąĄčüąĮąĖčģ ą┐čĆąŠąĖąĘą▓ąŠą┤ą░. ąĪčĆąĄą┤čÜąŠčłą║ąŠą╗čåąĖ ą▓ąĄčø ąĖą╝ą░čśčā ą▓ąĖčüąŠą║čā č鹊ą╗ąĄčĆą░ąĮčåąĖčśčā ąĮą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ, čéą░ą║ąŠ ą┤ą░ čā ąŠą▓ąŠčś čüčéą░čĆąŠčüčéąĖ ą╝ąŠąČąĄč鹥 ąĮą░čüčéą░ą▓ąĖčéąĖ ą┤ą░ ą┐čĆąŠčłąĖčĆčāčśąĄč鹥 ąĖčüčģčĆą░ąĮčā, ą┤ąŠą║ čśąĄ ąĮąĄąŠą┐čģąŠą┤ąĮąŠ ą┐čĆą░čéąĖčéąĖ ąŠą┤ą│ąŠą▓ąŠčĆ ąĮą░ čüą▓ąĄ ą┐čĆąŠą╝ąĄąĮąĄ čā ąĖčüčģčĆą░ąĮąĖ. ąĪą╗ąĄą┤ąĄčøąĄ ą╝ąĄč鹊ą┤ąĄ čüąĄ ą║ąŠčĆąĖčüč鹥 ąĘą░ ą┐čĆą░čøąĄčÜąĄ čüčéą░čÜą░ ą┤ąĄč鹥čéą░:
- ą┐čĆąŠčåąĄąĮą░ ąĮąĄčāčĆąŠą╗ąŠčłą║ąĖčģ ą┐ą░čĆą░ą╝ąĄčéą░čĆą░, ą┐čüąĖčģąŠą╗ąŠčłą║ąŠą│ čüčéą░čÜą░;
- ą║ąŠąĮčéčĆąŠą╗ą░ ą┐ą░čĆą░ą╝ąĄčéą░čĆą░ ąĄą╗ąĄą║čéčĆąŠąĄąĮčåąĄčäą░ą╗ąŠą│čĆą░ą╝ą░;
- ąŠą┤čĆąĄčÆąĖą▓ą░čÜąĄ ąĮąĖą▓ąŠą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░.
ąōčĆčāą┐ą┐ąĖ ą┐čĆąŠą┤čāą║č鹊ą▓ ąĮą░ ą¤ąÜąŻ
ąŻ ąĖčüčģčĆą░ąĮąĖ ą┐ą░čåąĖčśąĄąĮą░čéą░ čüą░ ą¤ąÜąŻ ąĘą░čśąĄą┤ąĮąŠ čüą░ ąĮąĄ-ą┐čĆąŠč鹥ąĖąĮąĖą╝ą░ą¤čĆąŠąĖąĘą▓ąŠą┤ąĖ ąŠą┤ čüą║čĆąŠą▒ą░ ąĖ č鹥čĆą░ą┐ąĄčāčéčüą║ąĄ ą╝ąĄčłą░ą▓ąĖąĮąĄ čéą░ą║ąŠčÆąĄ čāą║čÖčāčćčāčśčā ą┐čĆąŠąĖąĘą▓ąŠą┤ąĄ ą┐čĆąĖčĆąŠą┤ąĮąŠą│ ą┐ąŠčĆąĄą║ą╗ą░. ą¤čĆąĖą╗ąĖą║ąŠą╝ čüą░čüčéą░ą▓čÖą░čÜą░ ą╝ąĄąĮąĖčśą░ čéčĆąĄą▒ą░ čśą░čüąĮąŠ ąĖąĘčĆą░čćčāąĮą░čéąĖ ą║ąŠą╗ąĖčćąĖąĮčā ą║ąŠąĮąĘčāą╝ąĖčĆą░ąĮąŠą│ ą┐čĆąŠč鹥ąĖąĮą░ ąĖ ąĮąĄ ą┐čĆąĄą╗ą░ąĘąĖčéąĖ ą┐čĆąĄą┐ąŠčĆčāč湥ąĮčā ą┤ąŠąĘčā ą┤ąŠą║č鹊čĆą░. ąöą░ ą▒ąĖ čüąĄ ąĖčüą║čÖčāčćąĖą╗ąĖ č鹊ą║čüąĖčćąĮąĖ ąĄč乥ą║čéąĖ ąĮą░ ąŠčĆą│ą░ąĮąĖąĘą░ą╝, čĆą░ąĘą▓ąĖčśąĄąĮąĄ čüčā 3 ą╗ąĖčüč鹥 ą┐čĆąŠąĖąĘą▓ąŠą┤ą░ ą║ąŠčśąĖ čüą░ą┤čƹȹĄ ąĘą░ą▒čĆą░čÜąĄąĮąĄ (čåčĆą▓ąĄąĮąĄ), ąĮąĄą┐čĆąĄą┤čüčéą░ą▓čÖąĄąĮąĄ (ąĮą░čĆą░ąĮ褹░čüč鹥) ąĖ ą┤ąŠąĘą▓ąŠčÖąĄąĮąĄ (ąĘąĄą╗ąĄąĮąĄ) ą┐ąŠąĘąĖčåąĖčśąĄ.
ą”čĆą▓ąĄąĮą░ ą╗ąĖčüčéą░
ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čüąĄ čĆą░ąĘą▓ąĖčśą░ ąĮą░ ą┐ąŠąĘą░ą┤ąĖąĮąĖ ąŠą┤čüčāčüčéą▓ą░ ąĄąĮąĘąĖą╝ą░ ą║ąŠčśąĖ čüąĄ ą┐čĆąĄčéą▓ą░čĆą░ čā čéąĖčĆąŠąĘąĖąĮ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮ, čéą░ą║ąŠ ą┤ą░ čśąĄ ą▓ąĖčüąŠą║ čüą░ą┤čƹȹ░čś ą┐čĆąŠč鹥ąĖąĮą░ ąŠčüąĮąŠą▓ą░ ąĘą░ ą┤ąŠą┤čśąĄčÖąĖą▓ą░čÜąĄ ą┐čĆąŠąĖąĘą▓ąŠą┤ą░ ąĮą░ ąĘą░ą▒čĆą░čÜąĄąĮčā (čåčĆą▓ąĄąĮčā) ą╗ąĖčüčéčā. ą¤ąŠąĘąĖčåąĖčśąĄ čüą░ ąŠą▓ąĄ ą╗ąĖčüč鹥 čéčĆąĄą▒ą░ ą┤ą░ čā ą┐ąŠčéą┐čāąĮąŠčüčéąĖ ąĖčüą║čÖčāč湥 ąĖčüčģčĆą░ąĮčā ą┐ą░čåąĖčśąĄąĮčéą░ ąĮą░ ą¤ąÜąŻ:
- ą╝ąĄčüąŠ;
- čāąĮčāčéčĆą░čłčÜąĖ ąŠčĆą│ą░ąĮąĖ ąČąĖą▓ąŠčéąĖčÜą░, ąĮčāčüą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ;
- ą║ąŠą▒ą░čüąĖčåąĄ, ą║ąŠą▒ą░čüąĖčåąĄ;
- ą┐ą╗ąŠą┤ąŠą▓ąĖ ą╝ąŠčĆą░ (čāą║čÖčāčćčāčśčāčøąĖ čĆąĖą▒ąĄ);
- čśą░čśą░ čüą▓ąĖčģ ą┐čéąĖčåą░;
- ą┐čĆąŠąĖąĘą▓ąŠą┤ąĄ ąŠą┤ ą║ąĖčüąĄą╗ąŠą│ ą╝ą╗ąĄą║ą░;
- ą╝ą░čéąĖčåąĄ;
- ą╝ą░čģčāąĮą░čĆą║ąĄ ąĖ ąČąĖčéą░čĆąĖčåąĄ;
- ą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ ąŠą┤ čüąŠčśąĄ;
- ąČąĄą╗ą░čéąĖąĮąŠąĘąĮąĄ ą┐ąŠčüčāą┤ąĄ;
- ą║ąŠąĮą┤ąĖč鹊čĆčüą║ąĖ ą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ;
- ą░čüą┐ą░čĆčéą░ą╝.
ąØą░čĆą░ąĮ褹░čüčéą░ ą╗ąĖčüčéą░
ą¤čĆąŠąĖąĘą▓ąŠą┤ąĖ ą║ąŠčśąĖ čüąĄ čāąĮąŠčüąĄ čā č鹥ą╗ąŠ ą┤ąĄč鹥čéą░ čüą░ ą┤ąĖčśą░ą│ąĮąŠąĘąŠą╝ ą¤ąÜąŻ čāą║čÖčāč湥ąĮąĖ čüčā čā ąĮą░čĆą░ąĮ褹░čüčéčā ą╗ąĖčüčéčā. ąŻą║čÖčāčćąĖą▓ą░čÜąĄ čā ąĖčüčģčĆą░ąĮčā ą┐čĆąĄą┤ą╝ąĄčéą░ ąĖąĘ ąŠą▓ąĄ ą╗ąĖčüč鹥 čśąĄ ą┤ąŠąĘą▓ąŠčÖąĄąĮąŠ, ą░ą╗ąĖ čā čüčéčĆąŠą│ąŠ ąŠą│čĆą░ąĮąĖč湥ąĮąĖą╝ ą║ąŠą╗ąĖčćąĖąĮą░ą╝ą░. ąśą░ą║ąŠ ąŠą▓ąĖ ą┐čĆąŠąĖąĘą▓ąŠą┤ąĖ ąĮąĄ čüą░ą┤čƹȹĄ ą╝ąĮąŠą│ąŠ ą┐čĆąŠč鹥ąĖąĮą░, ą░ą╗ąĖ čśą░ čéą░ą║ąŠčÆąĄ ą╝ąŠą│čā ą┐ąŠą▓ąĄčøą░čéąĖ ąĮąĖą▓ąŠ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░, čüč鹊ą│ą░ čüąĄ čÜąĖčģąŠą▓ą░ čāą┐ąŠčéčĆąĄą▒ą░ ąĮąĄ ą┐čĆąĄą┐ąŠčĆčāčćčāčśąĄ:
- \ čé
- ą║ąŠąĮąĘąĄčĆą▓ąĖčĆą░ąĮąŠ ą┐ąŠą▓čĆčøąĄ;
- čśąĄą╗ą░ ąŠą┤ ą║čĆąŠą╝ą┐ąĖčĆą░ ąĖ čĆąĖąČąĄ;
- ą║čāą┐čāčü;
- ą╝ą╗ąĄą║ąŠ;
- ą©ąĄčĆą▒ąĄčé.
ąŚąĄą╗ąĄąĮą░ ą╗ąĖčüčéą░
- \ čé
- ą▓ąŠčøąĄ;
- ą┐ąŠą▓čĆčøąĄ (ąŠčüąĖą╝ ą║čĆąŠą╝ą┐ąĖčĆą░ ąĖ ą║čāą┐čāčüą░);
- ą▒ąŠą▒ąĖčåąĄ;
- ąĘąĄą╗ąĄąĮąĖ;
- čüą║čĆąŠą▒ąĮąĄ ąČąĖčéą░čĆąĖčåąĄ (čüą░ą│ąŠ);
- ą╝ąĄą┤, čłąĄčøąĄčĆ, 褹Ąą╝;
- ą▒čĆą░čłąĮąŠ ąŠą┤ ą║čāą║čāčĆčāąĘąĮąŠą│ ąĖą╗ąĖ ą┐ąĖčĆąĖąĮčćą░ąĮąŠą│ ą▒čĆą░čłąĮą░;
- ą┐čāč鹥čĆ, ą╝ą░čüčéąĖ (ą╝ą░čüą╗ą░čå, čüčāąĮčåąŠą║čĆąĄčé, ą╝ą░čüą╗ąĖąĮą░).
ąÜą░ą║ąŠ ą║ąŠąĮčéčĆąŠą╗ąĖčüą░čéąĖ ąĮąĖą▓ąŠ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā ą║čĆą▓ąĖ
ążąĄąĮąĖą╗ą║ąĄč鹊ąĮčāčĆąĖčśą░ čśąĄ ąĮąĄąĖąĘčÖąĄčćąĖą▓ą░ ą▒ąŠą╗ąĄčüčé ą║ąŠčśą░ čüąĄ ą╝ąŠąČąĄ ą┐čĆąĄčéą▓ąŠčĆąĖčéąĖ čā čäą░ąĘčā čüčéą░ą│ąĮą░čåąĖčśąĄ ą┐čĆąĖą╝čśąĄąĮąŠą╝ ą┤ąĖčśąĄč鹥čéčüą║ąĄ č鹥čĆą░ą┐ąĖčśąĄ ąĖ č鹥čĆą░ą┐ąĖčśčüą║ąĖčģ ąĖ ą┐čĆąŠčäąĖą╗ą░ą║čéąĖčćą║ąĖčģ ą╝čśąĄčĆą░. ąÜą░ą┤ą░ čüąĄ ą┐čĆąŠą╝ąĄąĮąĄ čāčüą╗ąŠą▓ąĖ ąČąĖą▓ąŠčéą░, ą║čĆčłąĄčÜąĄ ąĖčüčģčĆą░ąĮąĄ, ą▒ąŠą╗ąĄčüčé ą╝ąŠąČąĄ ą┐ąŠąĮąŠą▓ąŠ ą┤ą░ čüąĄ ą┐ąŠą│ąŠčĆčłą░, čéą░ą║ąŠ ą┤ą░ ą┐ą░čåąĖčśąĄąĮčéąĖą╝ą░ čéčĆąĄą▒ą░ ą┤ąŠąČąĖą▓ąŠčéąĮąŠ ą┐ąŠčüą╝ą░čéčĆą░čÜąĄ. ąÜąŠąĮčéčĆąŠą╗ąĮąĖ ą┐čĆąŠčåąĄčü čśąĄ ą┐ąĄčĆąĖąŠą┤ąĖčćąĮąŠ ąŠą┤čĆąĄčÆąĖą▓ą░čÜąĄ ąĮąĖą▓ąŠą░ č乥ąĮąĖą╗ą░ą╗ą░ąĮąĖąĮą░ čā ą║čĆą▓ąĖ. ąŻč湥čüčéą░ą╗ąŠčüčé ąĖčüą┐ąŠčĆčāą║ąĄ ąĘą░ą▓ąĖčüąĖ ąŠą┤ čüčéą░čĆąŠčüčéąĖ ą┐ą░čåąĖčśąĄąĮčéą░:
- \ čé
- ą┤ąŠ 3 ą╝ąĄčüąĄčåą░ - čüą║čĆąĖąĮąĖąĮą│ ą║čĆą▓ąĖ čéčĆąĄą▒ą░ čāčĆą░ą┤ąĖčéąĖ čśąĄą┤ąĮąŠą╝ ąĮąĄą┤ąĄčÖąĮąŠ ą┤ąŠą║ čüąĄ ąĮąĄ ą┤ąŠą▒ąĖčśčā čüčéą░ą▒ąĖą╗ąĮąĖ čĆąĄąĘčāą╗čéą░čéąĖ;
- ąŠą┤ 3 ą╝ąĄčüąĄčåą░ ą┤ąŠ 1 ą│ąŠą┤ąĖąĮąĄ - 1-2 ą┐čāčéą░ ą╝ąĄčüąĄčćąĮąŠ;
- ąŠą┤ 1 ą┤ąŠ 3 ą│ąŠą┤ąĖąĮąĄ - 1 ą┐čāčé čā 2 ą╝ąĄčüąĄčåą░;
- ą┐čĆąĄą║ąŠ 3 ą│ąŠą┤ąĖąĮąĄ - ą║ą▓ą░čĆčéą░ą╗ąĮąŠ.

ąÆąĖą┤ąĄąŠ
ąśąĮč乊čĆą╝ą░čåąĖčśąĄ čā čćą╗ą░ąĮą║čā čüčā ąĖąĮč乊čĆą╝ą░čéąĖą▓ąĮąĄ. ą£ą░č鹥čĆąĖčśą░ą╗ąĖ čā ąŠą▓ąŠą╝ čćą╗ą░ąĮą║čā ąĮąĄ ąĘą░čģč鹥ą▓ą░čśčā ąĮąĄąĘą░ą▓ąĖčüąĮąĖ čéčĆąĄčéą╝ą░ąĮ. ąĪą░ą╝ąŠ ą║ą▓ą░ą╗ąĖčäąĖčåąĖčĆą░ąĮąĖ ą╗ąĖčśąĄčćąĮąĖą║ ą╝ąŠąČąĄ ą┤ąĖčśą░ą│ąĮąŠčüčéąĖčåąĖčĆą░čéąĖ ąĖ ą┤ą░čéąĖ čüą░ą▓čśąĄč鹥 ąŠ ą╗ąĖčśąĄč湥čÜčā ąĮą░ č鹥ą╝ąĄčÖčā ąĖąĮą┤ąĖą▓ąĖą┤čāą░ą╗ąĮąĖčģ ą║ą░čĆą░ą║č鹥čĆąĖčüčéąĖą║ą░ ąŠą┤čĆąĄčÆąĄąĮąŠą│ ą┐ą░čåąĖčśąĄąĮčéą░.








